
Fluorous solid-phase extraction (F-SPE) as a pilot tool for quantitative determination of perfluorochemicals in water samples coupled with liquid chromatography-tandem mass spectrometry
Abstract
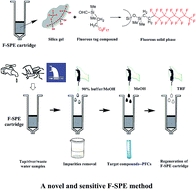
As a large group of stable existing organofluorine compounds widely present in the environment, perfluorochemicals (PFCs) could pose potential adverse effects on human health. In this study, a selective and sensitive fluorous solid-phase extraction (F-SPE) method coupled with liquid chromatography-tandem mass spectrometry (LC-MS/MS) was developed and validated for quantitative analysis of PFCs. The novel fluorous solid-phase cartridges used in the present work were synthesized, which have a fluorous solid phase being typically silica gel with a fluorocarbon bonded phase (–SiMe2(CH2)2C8F17). As a result of the fluorous–fluorous interaction, F-SPE displayed excellent extraction efficiency for the PFCs, including perfluorohexanoic acid (PFHxA), perfluorooctanoic acid (PFOA), perfluorononanoic acid (PFNA), and perfluorodecanoic acid (PFDA). The method contains 4 steps: preconditioning, sample loading, fluorophobic washing and fluorophilic elution. Meanwhile, the cartridges could be regenerated by thorough washing with tetrahydrofuran (THF) and reused multiple times. The analytes collected from F-SPE were redissolved and injected into the LC-MS/MS system for quantitation afterwards. The separation was performed in negative ion mode using multiple reaction monitoring mode on a Shiseido C18 column (150 × 4.6 mm, 5.0 μm) using methanol and buffer (2 mM ammonium acetate, 0.05% HAc) as mobile phases with a gradient of 80/20 (v/v). The four kinds of PFCs could be eluted within 13 min with analytical recoveries being in the range of 95.3–102.8%. The developed method was applied to determine the concentration of PFCs in tap/river/waste samples collected in Shanghai, China which indicates the prospective value of F-SPE for environmental monitoring and protection.
 Please wait while we load your content...
Please wait while we load your content...

