
Interspecies prediction of oral pharmacokinetics of different lacidipine formulations from dogs to human: physiologically based pharmacokinetic modelling combined with biorelevant dissolution
Abstract
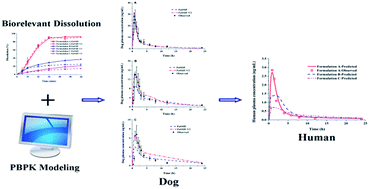
The aim of the present study was to use physiologically based pharmacokinetic (PBPK) modelling combined with biorelevant dissolution to quantitatively predict dog oral drug pharmacokinetic (PK) of different formulations, and to make an interspecies extrapolation to human. Lacidipine, a typical BCS class II drug, was chosen as a model drug. The three lacidipine formulations involved were immediate release tablets including two commercial products and one self-made micronized tablets. First of all, the PBPK models in rat and dog after intravenous administration of lacidipine solutions and oral administration of lacidipine suspensions were developed and validated based on physicochemical properties and the literature data, which were used to verify the accuracy of the PBPK model in prediction drug PK in animals. The solubility and dissolution behaviour of lacidipine formulations were determined in the media simulating the fasted state intestinal fluid of human (FaSSIF/FaSSIF-V2). Then the Z-factor values obtained by fitting the dissolution profiles of the three formulations were utilized to evaluate the effect of in vitro dissolution behaviours on PK performance. At last, the verified PBPK model in dog was extrapolated to predict human PK of the three formulations. The results showed that PBPK models from dog to human were successfully developed. By integrating biorelevant dissolution with PBPK model, it is achievable to quantitatively and accurately predict the plasma concentration–time profiles for various formulations after oral administration.
 Please wait while we load your content...
Please wait while we load your content...

