
Understanding the binding interaction of imidazole with ZnO nanomaterials and clusters†
Abstract
The potential applications of different nanostructured materials in biomedical nanotechnologies and the interaction of nanomaterials with bioactive molecules are of current interest. A bioactive fluorophore 1-(3,5-dimethylphenyl)-2-(furan-2-yl)-4,5-diphenyl-1H-imidazole has been synthesized and characterized by 1H NMR, 13C NMR and mass spectral studies. Electronic properties and the binding interactions of the fluorophore with ZnO nanocrystals of different sizes have been studied using absorption, emission, lifetime and cyclic voltammetric analysis. The strong adsorption of the imidazole on the surface of ZnO nanocrystals is likely due to the chemical affinity of azomethine nitrogen atom of the imidazole and this is likely to result in lowering the HOMO and LUMO energy levels. TEM, SEM and EDS confirm the adsorption of imidazole on the surface of ZnO nanocrystals. Theoretical investigation reveals that small ZnnOn clusters (n < 9) and their imidazole–ZnO composites are stabilized in 2D ring geometries whereas the larger cluster Zn10O10 and its imidazole–ZnO composite prefer 3D cage structures. The ring to cage crossover of ZnO clusters is studied by analyzing the 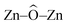 and
and 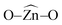 bond angles, Zn–O bond length and number of bonds. Binding energy, energy gap, binding site and adsorption strength of imidazole with different ZnO clusters show that Zn2+ of ZnO clusters prefer to bind with the azomethine nitrogen atom (N-site) relative to the other binding site (O-site).
bond angles, Zn–O bond length and number of bonds. Binding energy, energy gap, binding site and adsorption strength of imidazole with different ZnO clusters show that Zn2+ of ZnO clusters prefer to bind with the azomethine nitrogen atom (N-site) relative to the other binding site (O-site).

 Please wait while we load your content...
Please wait while we load your content...

