
Efficient charge transport of a radical polyether/SWCNT composite electrode for an organic radical battery with high charge-storage density†
Takashi Sukegawa,
Kan Sato,
Kenichi Oyaizu* and
Hiroyuki Nishide*
Department of Applied Chemistry, Waseda University, Tokyo 169-8555, Japan. E-mail: nishide@waseda.jp; Fax: +81-3-3209-5522; Tel: +81-3-3200-2669
First published on 26th January 2015
Abstract
A crosslinked poly(4-glycidyloxy TEMPO) was synthesized via anionic ring-opening copolymerization of 4-glycidyloxy TEMPO and a diglycidyl ether using a pentaerythritol/phosphazene base initiator. A fast and reversible charge storage capability was established for the polyether/SWCNT composite layer with a large layer thickness of several tens of micrometres, despite the low SWCNT content of 10% which was much closer to the percolation limit than the amount required for the previously reported TEMPO-substituted polymethacrylate.
Chemistry of charge storage with electrode-active materials is dominated by a mass-transfer process of electrolyte ions to accomplish charge compensation throughout the electroactive layer.1–3 The electroneutralization process is much influenced by diffusivity of the counterion, which has prompted many studies on electrode-active materials designed for high ionic conductivity, such as mesostructured lithium metalates4–11 and polyethers bearing redox-active pendant groups.12 As typical organic ion conductors, poly(ethylene oxide) (PEO)-based materials have attracted much attention for their potential application in electronic devices, such as light-emitting diodes,13,14 thin-film transistors,15–17 sensors,18 solar cells,19,20 and Li-ion batteries,21–24 which are expected to acquire unprecedented properties related to flexibility, light weight, and safety. In particular, a PEO-based electrolyte layer has been investigated as a Li+ single conductor for many types of rechargeable devices.25–27 High ionic conductivity for the electrode-active layer is also crucial for charge compensation. Incorporation of PEO-based materials has therefore been widely examined to enhance the charging/discharging performance.28–32 For example, a multifunctional block copolymer containing a PEO segment and a conductive polymer segment was proposed as an ionic and electronic (or mixed) conductor and a binder within the cathode layer.33,34
We have been pursuing organic radical-containing polymers as electrode-active materials in organic secondary batteries which are characterized by the excellent electrochemical properties such as the high charging/discharging rate and the long cycle stability.35–40 Charge transport through a layer of aliphatic polymers populated with the organic robust radicals per repeating unit, or radical polymers, consist of an electron self-exchange reaction between the redox-active radical sites and the electroneutralization with counterions. Among them, radical polyethers containing nitroxides such as 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) and 2,2,5,5-tetramethylpyrrolidin-1-oxyl enabled quantitative charging/discharging at high current densities and long cycle life which was most likely attributed to the nature of the main chain analogous to PEO allowing ionic transport throughout the polymer layer and sufficient swelling in conventional electrolyte solutions. The PEO chain was also characterized by the excellent chemical stability against the redox reaction, allowing charging/discharging for more than 1000 cycles.12,41 The radical polyether is expected to contribute to increase the thickness of the polymer layer and yet to reduce the amount of carbon nanofibers as electroconductive additives while maintaining the rate performance of the polymer/carbon composite electrode, thus giving rise to larger charge-storage density of the composite layer compared with other radical polymers.
However, inherent difficulties in the synthesis of radical polyethers have deprived them of such opportunities. The radical polyethers were originally synthesized via anionic ring-opening polymerizations of radical monomers using potassium tert-butoxide (t-BuOK) or diethylzinc/H2O catalyst. These polymerizations resulted in either very low yields and/or deficits of radicals which primarily decreased the charging capacity of the organic batteries.42,43
In this paper, we report a controlled synthesis of crosslinked poly(4-glycidyloxy TEMPO) (CL-PTGE) with high molecular weights and radical concentrations, and an efficient charge transport of remarkable polymer-rich CL-PTGE/carbon composite electrodes with large thicknesses up to several ten micrometers maintaining the redox capacity. Our principal finding was that alcohols with a phosphazene base44,45 was an excellent initiator for the anionic ring-opening polymerization of 4-glycidyloxy TEMPO, compared with t-BuOK which was the most conventional for glycidyl monomers. The present initiator allowed many types of polyether architectures according to the structure of the alcohols or polyols, which led to good dispersion of a single-walled carbon nanotube (SWCNT) in the polymer.46,47 The CL-PTGE composite electrode demonstrated the fast and quantitative charging/discharging even in the thick layers which several ten micrometers in thickness.
4-Glycidyloxy TEMPO was synthesized via the Williamson ether synthesis from epichlorohydrin and 4-hydroxy-TEMPO. PTGE was prepared at room temperature via the anionic ring-opening polymerization of 4-glycidyloxy TEMPO using 3-phenyl-1-propanol (ROH)/tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis[tris(dimethylamino)phosphoranylidenamino]-2Λ5,4Λ5-catenadi(phosphazene) (t-Bu-P4), a strong base with low nucleophilicity, or pentaerythritol (R(OH)4)/t-Bu-P4 as a monofunctional and a quadruply-branched initiator, respectively (Scheme 1). The moderate temperature was required to suppress undesirable side reactions involving a deactivation of the nitroxide radical moiety as observed for the t-BuOK initiator that required higher temperatures above 60 °C.41 The results of the polymerization are summarized in Table S1.† PTGE was obtained with a low polydispersity index (PDI) in ca. 70% yield using the ROH/t-Bu-P4 initiator at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, while the molecular weight was lower than the theoretical value based on the initial composition ratio of [monomer]/[initiator] = 100/1. The R(OH)4/t-Bu-P4 initiator at a molar ratio of 1:4 ([OH]/[t-Bu-P4] = 1/1) produced PTGE with the narrow PDI in a significantly higher yield of 90%. Furthermore, the R(OH)4/t-Bu-P4 initiator at a molar ratio of 1:2 ([OH]/[t-Bu-P4] = 2/1) gave PTGE in 80% yield, in contrast to the R(OH)/t-Bu-P4 initiator at the same molar ratio of [OH]/[t-Bu-P4] which produced only a trace amount of the polymer in 4% yield. Copolymerization of 4-glycidyloxy TEMPO and 1,4-butanediol diglycidyl ether as a crosslinker was carried out to yield the CL-PTGE with a sufficient crosslinking density to realize the swelling but insoluble properties in organic electrolyte solutions. The CL-PTGE was obtained with the R(OH)4/t-Bu-P4 initiator at [monomer]/[crosslinker] = 100/5 in 98% yield which was increased from the yield of 80% for the corresponding homopolymerization, while the copolymerization using ROH/t-Bu-P4 resulted in only 77% yield. The CL-PTGE contained low composition of the redox-inactive crosslinker to minimize the loss of the charge capacity. This efficient crosslinking was attributed to the high reactivity of the alkoxide anion activated by t-Bu-P4 and the quadruply-branched structure of R(OH)4 which also served as the crosslinking point. The unpaired electron density of 0.92 per monomer unit for CL-PTGE, estimated by the SQUID magnetic measurements, revealed that the radical survived during the polymerization by virtue of the low nucleophilicity of t-Bu-P4 and the moderate reaction temperature (Fig. S1†).
:1, while the molecular weight was lower than the theoretical value based on the initial composition ratio of [monomer]/[initiator] = 100/1. The R(OH)4/t-Bu-P4 initiator at a molar ratio of 1:4 ([OH]/[t-Bu-P4] = 1/1) produced PTGE with the narrow PDI in a significantly higher yield of 90%. Furthermore, the R(OH)4/t-Bu-P4 initiator at a molar ratio of 1:2 ([OH]/[t-Bu-P4] = 2/1) gave PTGE in 80% yield, in contrast to the R(OH)/t-Bu-P4 initiator at the same molar ratio of [OH]/[t-Bu-P4] which produced only a trace amount of the polymer in 4% yield. Copolymerization of 4-glycidyloxy TEMPO and 1,4-butanediol diglycidyl ether as a crosslinker was carried out to yield the CL-PTGE with a sufficient crosslinking density to realize the swelling but insoluble properties in organic electrolyte solutions. The CL-PTGE was obtained with the R(OH)4/t-Bu-P4 initiator at [monomer]/[crosslinker] = 100/5 in 98% yield which was increased from the yield of 80% for the corresponding homopolymerization, while the copolymerization using ROH/t-Bu-P4 resulted in only 77% yield. The CL-PTGE contained low composition of the redox-inactive crosslinker to minimize the loss of the charge capacity. This efficient crosslinking was attributed to the high reactivity of the alkoxide anion activated by t-Bu-P4 and the quadruply-branched structure of R(OH)4 which also served as the crosslinking point. The unpaired electron density of 0.92 per monomer unit for CL-PTGE, estimated by the SQUID magnetic measurements, revealed that the radical survived during the polymerization by virtue of the low nucleophilicity of t-Bu-P4 and the moderate reaction temperature (Fig. S1†).
 | ||
| Scheme 1 Anionic ring-opening copolymerization of 4-glycidyloxy TEMPO and 1,4-butanediol diglycidyl ether. | ||
SWCNT with 1–3 nm in diameter was incorporated into a CL-PTGE layer to support long-range conduction throughout the layer, with a view to maximize the overall energy density of the battery by reducing the amount of the conductive additive down to approach the theoretically predicted percolation limit of less than 1%.48–50 This has been an important issue in the studies of organic electrode-active materials, but has never been accomplished with the conventional redox polymer/vapor-grown carbon nanofiber (VGCF) composites which always required a VGCF composition of 40–60 wt%.51,52 CL-PTGE and SWCNT were uniformly dispersed in CHCl3, a good solvent for both materials, using a probe sonicator and then the resultant mixture was cast-coated on a current collector. A SEM image of the composite electrode indicated that 10 wt% SWCNT was homogeneously buried in a 90 wt% CL-PTGE matrix and formed a network in a micrometer scale despite that SWCNT still formed bundles (Fig. S2(a)†). A smaller amount of SWCNT (5 wt%) was contrastingly buried in the 95 wt% CL-PTGE matrix without forming the network (Fig. S2(b)†). A much lower-density network was formed with 10 wt% VGCF with a larger diameter of 150 nm than that of SWCNT (Fig. S2(c)†). The higher dispersivity of SWCNT obviously suggested the affinity with PTGE, which was reminiscent of the molecular wrapping of SWCNT with a TEMPO-substituted polymethacrylate in our previous study.47 The conductivity of the 10 wt% SWCNT composite (42 mS cm−1), measured by the four-probe method with 1 mm regular probe intervals, was higher than those of the 5 wt% SWCNT composite (14 mS cm−1) and the 10 wt% VGCF composite (2.3 mS cm−1) (Table S2†), suggesting that the 10 wt% SWCNT was sufficient to form the conductive path throughout the composite electrode at the macroscopic scale.
Electrochemical properties of the three composite electrodes with ca. 10 μm in thickness were examined in a typical three-electrode cell using a Pt coil and an Ag/AgCl as the counter and the reference electrodes, respectively. Cyclic voltammograms obtained with the composite electrodes all displayed a reversible redox wave at 0.73 V (vs. Ag/AgCl) in a 0.1 M (n-C4H9)4NClO4/acetonitrile electrolyte solution (Fig. 1 and S3†).
 | ||
| Fig. 1 (a) Cyclic voltammograms obtained for the CL-PTGE electrodes containing 0 or 10 wt% SWCNT at a scan rate of 5 mV s−1. The electrolyte was a solution of 0.1 M (n-C4H9)4NClO4 in CH3CN. (b) Schematic representation for charge transportation in the CL-PTGE/SWCNT composite layer swelling the electrolyte solution. | ||
The redox cyclability of the electrodes indicated that CL-PTGE strongly adhered to the electrode substrate and hence served also as a binder. The 10 wt% SWCNT composite electrode displayed a narrow peak-to-peak separation and a large charging capacity in comparison with the 0 and 5 wt% SWCNT and the 10 wt% VGCF composites. Coulometric titration experiments by chronopotentiometry also revealed the highest charging/discharging capacity for the 10 wt% SWCNT composite electrode (Fig. S4†).
The excellent charge storage properties of the 10 wt% SWCNT composite electrode originated from the higher conductivity of the layer. The charging/discharging properties of the 5 wt% and the 10 wt% SWCNT composite electrodes and the non-composite (or pristine polymer-coated) electrode with ca. 40 μm in thickness were examined (Fig. 2(a)). The 10 wt% SWCNT markedly increased the discharging capacity of the composite electrode up to 93 mA g−1 that corresponded to 90% of the theoretical or formula weight-based capacity from 20 mA h g−1 (20%) for the non-composite electrode, while the charging capacity of the 5 wt% SWCNT composite electrode was limited to 69 mA h g−1. The difference in plateau potentials for discharging between 0.62 V for the 10 wt% SWCNT composite electrode and 0.54 V for the non-composite electrode demonstrated the less resistive properties established with SWCNT. The 10 wt% SWCNT composite electrode displayed a high current density, even at a 30 C rate, with a quantitative charging/discharging capacity (Fig. 2(b)).‡ The resistance of the composite electrodes were also estimated from AC impedance measurements. The total charge-transfer resistance for the 10 wt% SWCNT composite electrode (41 Ω cm−2) was comparable to that of the non-composite electrode of CL-PTGE (44 Ω cm−2), while other composite electrodes exhibited higher resistances (Table S2†). Transition from the diffusion controlled region (with a slope of 45° to the x axis) to the charge saturation region (90°) in the impedance spectrum for the 10 wt% SWCNT composite electrode indicated that the finite diffusion was realized at low frequencies, while the non-composite CL-PTGE exhibited a semi-infinite diffusional behavior even at very low frequency regions (Fig. S5†). This difference was most likely ascribed to the shortened diffusion length for the electroneutralizing ions in the composite layer due to the long-range charge transport supported by the SWCNT network throughout the composite layer.
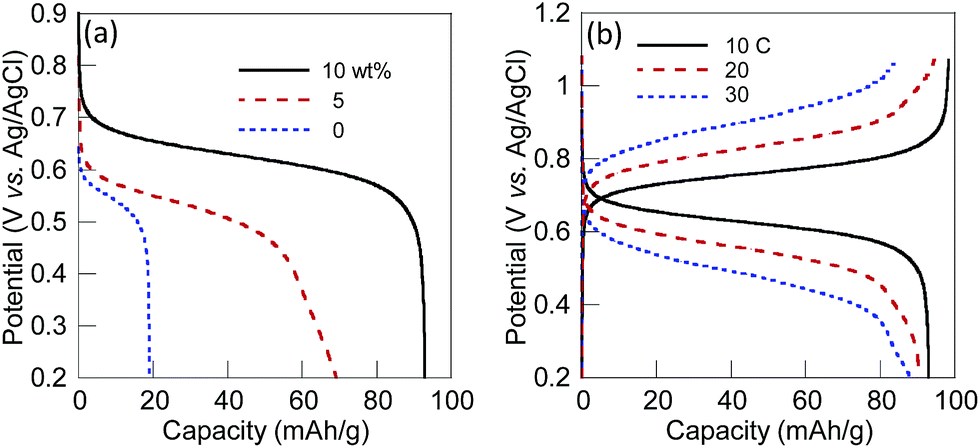 | ||
| Fig. 2 Charging/discharging performance of the CL-PTGE electrodes (a) containing 0, 5, and 10 wt% SWCNT at 10 C rate, and (b) containing 10 wt% SWCNT at 10–30 C rate. The electrolyte was a solution of 0.1 M (n-C4H9)4NClO4 in CH3CN. | ||
We fabricated a test full-cell consisted of a 10 wt% SWCNT composite cathode, a lithium anode, a separator film, and a 1.0 M LiPF6 in ethylene carbonate/diethyl carbonate electrolyte solution. The charging/discharging curve at 1 C rate displayed a plateau voltage at 3.5 V corresponding to the potential difference between the cathode and the anode (Fig. 3).‡ The charge capacity was 80 mA h g−1 (80% of the theoretical capacity) and maintained over 70% of the initial capacity after 200 cycles. The excellent rate performance for the charging/discharging process of the CL-PTGE composite electrode gave rise to a high rate capability of the cell, allowing fast charging and discharging in 10 C without loss of the charge-storage capacity. Whilst some inorganic electrode-active materials in lithium ion batteries have been recently achieving the fast charging/discharging capabilities (>100 C),53,54 organic electrode-active materials still have advantages in terms of tuning the chemical structures for faster ion mobilities and quick redox reactions allowing the ultrafast charging/discharging ability (>2000 C).55 Fabricating the high-performance, flexible, and lightweight all-organic batteries56 will also be our next challenge.
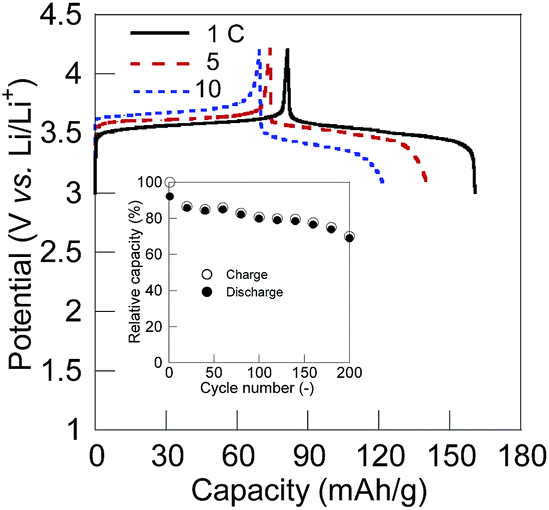 | ||
| Fig. 3 Charging/discharging curves for a coin-type cell consisted of the CL-PTGE/10 wt% SWCNT composite cathode and the lithium anode with 1.0 M LiPF6 in ethylene carbonate/diethyl carbonate (1/1 in v/v) at 1–10 C rates. Inset: cycle performance of the cell. | ||
Conclusions
CL-PTGE was successfully synthesized via anionic ring-opening copolymerization of the 4-glycidyloxy TEMPO and a small amount of the diglycidyl ether as a crosslinker using pentaerythritol/t-Bu-P4 initiator. The CL-PTGE/SWCNT composite electrode (9/1 in w/w) exhibited a quantitative charging/discharging at high current densities with a long cycle life, due to the low resistance supported by the conductive network of SWCNT and the high affinity of CL-PTGE to the electrolyte solution. The combination of the crosslinked poly(glycidyl ether) and SWCNT showed great potential in utilizing redox polymers for energy devices.Acknowledgements
This work was partially supported by Grants-in-Aid for Scientific Research (no. 24108739, 24225003, 25107733, and 25288056) and the Leading Graduate Program in Science and Engineering, Waseda University from MEXT, Japan. Support by the project “Functional Redox Polymers” of Waseda Advanced Research Institute for Science & Engineering is acknowledged.Notes and references
- H. Nishide and K. Oyaizu, Science, 2008, 319, 737–738 CrossRef CAS PubMed
.
- K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 2339–2344 CrossRef CAS
- W. Choi, D. Harada, K. Oyaizu and H. Nishide, J. Am. Chem. Soc., 2011, 133, 19839–19843 CrossRef CAS PubMed
- P. L. Taberna, S. Mitra, P. Poizot, P. Simon and J.-M. Tarascon, Nat. Mater., 2006, 5, 567–573 CrossRef CAS PubMed
- A. Manthiram and J. Kim, Chem. Mater., 1998, 10, 2895–2909 CrossRef CAS
- M. Wilkening, C. Mühle, M. Jansen and P. Heitjans, J. Phys. Chem. B, 2007, 111, 8691–8694 CrossRef CAS PubMed
- G. Yang, G. Wang and W. Hou, J. Phys. Chem. B, 2005, 109, 11186–11196 CrossRef CAS PubMed
- P. Zhang, Y. Wu, D. Zhang, Q. Xu, J. Liu, X. Ren, Z. Luo, M. Wang and W. Hong, J. Phys. Chem. A, 2008, 112, 5406–5410 CrossRef CAS PubMed
- A. S. Aricò, P. Bruce, B. Scrosati, J. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed
- S. Myung, S. Komaba, K. Hosoya, N. Hirosaki, Y. Miura and N. Kumagai, Chem. Mater., 2005, 17, 2427–2435 CrossRef CAS
- K. T. Nam, D. Kim, P. J. Yoo, C.-Y. Chiang, N. Meethong, P. T. Hammond, Y. Chiang and A. M. Belcher, Science, 2006, 312, 885–888 CrossRef CAS PubMed
- K. Oyaizu, T. Kawamoto, T. Suga and H. Nishide, Macromolecules, 2010, 43, 10382–10389 CrossRef CAS
- S. van Reenen, P. Matyba, A. Dzwilewski, R. A. J. Janssen, L. Edman and M. Kemerink, J. Am. Chem. Soc., 2010, 132, 13776–13781 CrossRef CAS PubMed
- T. Wågberg, P. R. Hania, N. D. Robinson, J.-H. Shin, P. Matyba and L. Edman, Adv. Mater., 2008, 20, 1744–1749 CrossRef
- J. Lee, M. J. Panzer, Y. He, T. P. Lodge and C. D. Frisbie, J. Am. Chem. Soc., 2007, 129, 4532–4533 CrossRef CAS PubMed
- K. H. Lee, S. Zhang, Y. Gu, T. P. Lodge and C. D. Frisbie, ACS Appl. Mater. Interfaces, 2013, 5, 9522–9527 CAS
- J. Liu, I. Engquist, X. Crispin and M. Berggren, J. Am. Chem. Soc., 2012, 134, 901–904 CrossRef CAS PubMed
- M. Yuasa and K. Oyaizu, Curr. Org. Chem., 2005, 9, 1685–1697 CrossRef CAS
- J. E. Benedetti, M. A. de Paoli and A. F. Nogueira, Chem. Commun., 2008, 1121–1123 RSC
- F. S. Freitas, J. N. de Freitas, B. I. Ito, M.-A. De Paoli and A. F. Nogueira, ACS Appl. Mater. Interfaces, 2009, 1, 2870–2877 CAS
- K. P. Barteau, M. Wolffs, N. A. Lynd, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Macromolecules, 2013, 46, 8988–8994 CrossRef CAS
- a. Blazejczyk, W. Wieczorek, R. Kovarsky, D. Golodnitsky, E. Peled, L. G. Scanlon, G. B. Appetecchi and B. Scrosati, J. Electrochem. Soc., 2004, 151, A1762 CrossRef CAS PubMed
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Lienafa, J. Bonnet, T. N. T. Phan, D. Bertin, D. Gigmes, D. Devaux, R. Denoyel and M. Armand, Nat. Mater., 2013, 12, 452–457 CrossRef CAS PubMed
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456–458 CrossRef CAS PubMed
- K. Sinha and J. K. Maranas, Macromolecules, 2011, 44, 5381–5391 CrossRef CAS
- K. Sinha, W. Wang, K. I. Winey and J. K. Maranas, Macromolecules, 2012, 45, 4354–4362 CrossRef CAS
- P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–282 CrossRef PubMed
- Z. Jia, W. Yuan, H. Zhao, H. Hu and G. L. Baker, RSC Adv., 2014, 4, 41087–41098 RSC
- H. Hu, W. Yuan, L. Lu, H. Zhao, Z. Jia and G. L. Baker, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2104–2110 CrossRef CAS
- H. Hu, W. Yuan, Z. Jia and G. L. Baker, RSC Adv., 2014, 5, 3135–3140 RSC
- M. J. Lacey, F. Jeschull, K. Edström and D. Brandell, Chem. Commun., 2013, 49, 8531–8533 RSC
- J. Xiao, X. Wang, X.-Q. Yang, S. Xun, G. Liu, P. K. Koech, J. Liu and J. P. Lemmon, Adv. Funct. Mater., 2011, 21, 2840–2846 CrossRef CAS
- A. E. Javier, S. N. Patel, D. T. Hallinan, V. Srinivasan and N. P. Balsara, Angew. Chem., Int. Ed. Engl., 2011, 50, 9848–9851 CrossRef CAS PubMed
- S. N. Patel, A. E. Javier and N. P. Balsara, ACS Nano, 2013, 7, 6056–6068 CrossRef CAS PubMed
- T. Suga, S. Sugita, H. Ohshiro, K. Oyaizu and H. Nishide, Adv. Mater., 2011, 23, 751–754 CrossRef CAS PubMed
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS PubMed
- I. S. Chae, M. Koyano, T. Sukegawa, K. Oyaizu and H. Nishide, J. Mater. Chem. A, 2013, 1, 9608 CAS
- T. Sukegawa, A. Kai, K. Oyaizu and H. Nishide, Macromolecules, 2013, 46, 1361–1367 CrossRef CAS
- L. Bugnon, C. J. H. Morton, P. Novak, J. Vetter and P. Nesvadba, Chem. Mater., 2007, 19, 2910–2914 CrossRef CAS
- T. Hyakutake, J. Y. Park, Y. Yonekuta, K. Oyaizu, H. Nishide and R. Advincula, J. Mater. Chem., 2010, 20, 9616 RSC
- T. Suga, K. Yoshimura and H. Nishide, Macromol. Symp., 2006, 245–246, 416–422 CrossRef
- T. Endo, K. Takuma, T. Takata and C. Hirose, Macromolecules, 1993, 26, 3227–3229 CrossRef CAS
- K. Oyaizu, T. Suga, K. Yoshimura and H. Nishide, Macromolecules, 2008, 41, 6646–6652 CrossRef CAS
- T. Isono, K. Kamoshida, Y. Satoh, T. Takaoka, S. Sato, T. Satoh and T. Kakuchi, Macromolecules, 2013, 46, 3841–3849 CrossRef CAS
- H. Misaka, E. Tamura, K. Makiguchi, K. Kamoshida, R. Sakai, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1941–1952 CrossRef CAS
- W. Choi, S. Endo, K. Oyaizu, H. Nishide and K. E. Geckeler, J. Mater. Chem. A, 2013, 1, 2999 CAS
- W. Choi, S. Ohtani, K. Oyaizu, H. Nishide and K. E. Geckeler, Adv. Mater., 2011, 23, 4440–4443 CrossRef CAS PubMed
- A. V. Kyrylyuk and P. van der Schoot, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8221–8226 CrossRef CAS PubMed
- J.-M. Benoit, B. Corraze, S. Lefrant, W. J. Blau, P. Bernier and O. Chauvet, Synth. Met., 2001, 121, 1215–1216 CrossRef CAS
- J. C. Grunlan, a. R. Mehrabi, M. V. Bannon and J. L. Bahr, Adv. Mater., 2004, 16, 150–153 CrossRef CAS
- S. Yoshihara, H. Katsuta, H. Isozumi, M. Kasai, K. Oyaizu and H. Nishide, J. Power Sources, 2011, 196, 7806–7811 CrossRef CAS PubMed
- S. Yoshihara, H. Isozumi, M. Kasai, H. Yonehara, Y. Ando, K. Oyaizu and H. Nishide, J. Phys. Chem. B, 2010, 114, 8335–8340 CrossRef CAS PubMed
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed
- W. Tang, S. Tian, L. L. Liu, L. Li, H. P. Zhang, Y. B. Yue, Y. Bai, Y. P. Wu and K. Zhu, Electrochem. Commun., 2011, 13, 205–208 CrossRef CAS PubMed
- K. Koshika, N. Sano, K. Oyaizu and H. Nishide, Chem. Commun., 2009, 836–838 RSC
- T. Suga, H. Ohshiro, S. Sugita, K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 1627–1630 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic method, electrode preparation, and electrochemical and SQUID measurements. See DOI: 10.1039/c4ra15949g |
| ‡ The 1 C rate is defined as the current density at which the charging or discharging of the cell takes 1 h. |
| This journal is © The Royal Society of Chemistry 2015 |