
Cytotoxicity, genotoxicity and alteration of cellular antioxidant enzymes in silver nanoparticles exposed CHO cells
Abstract
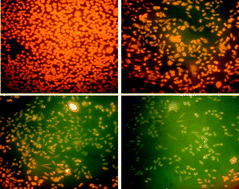
The broad applications of silver nanoparticles (Ag NPs) increase human exposure and thus the potential risk associated with their toxicity; therefore, the toxicity of Ag NPs, synthesized by a chemical route was studied using Chinese Hamster Ovary (CHO) cells. A UV-vis absorption maximum at 406 nm confirmed the formation of silver nanoparticles. The average diameter of the silver nanoparticles having spherical shape was found to be about 10.0 ± 1.0 nm by transmission electron microscopy (TEM). For toxicity evaluation, cellular morphology, mitochondrial function (MTT assay), mitochondrial membrane potential, anti-oxidant enzyme assay and comet assay were assessed in CHO cells exposed to various dose concentrations of 25, 50 and 100 μg ml−1 as well as in control cells. Ag NPs caused decreased mitochondrial membrane potential and comparable CAT, SOD, GPx, GST, GR activities as well as total Glutathione level. Comet tail length, tail moment and percent DNA in tail were also found to be increased in a dose dependent manner. In summary, the results suggest that Ag NPs of smaller size at low concentration (25 μg ml−1) cause cytotoxicity by oxidative stress induced apoptosis and damage to DNA and other cellular components.
 Please wait while we load your content...
Please wait while we load your content...

