
Synthesis of pillar[n]arenes (n = 5 and 6) with deep eutectic solvent choline chloride 2FeCl3†
Jin Caoa,
Yuhan Shanga,
Bin Qia,
Xuzhuo Sunb,
Lei Zhanga,
Huiwen Liua,
Haibo Zhang*a and
Xiaohai Zhoua
aCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan, 430072, China. E-mail: haibozhang1980@gmail.com
bCollege of Chemistry and Chemical Engineering, Henan University of Technology, Zhengzhou, 450001, China
First published on 5th January 2015
Abstract
A novel and distinct method of preparing pillar[5]arene and pillar[6]arene with high selectivity and efficiency has been achieved by condensation of 1,4-dialkoxybenzene and paraformaldehyde with the choline chloride (ChCl)/ferric chloride (FeCl3) deep eutectic solvent in CH2Cl2 at room temperature. Under the optimal conditions, the yield of pillar[5]arene and pillar[6]arene is 35% and 53%, respectively. The reaction mechanism is investigated by room-temperature X-band Electron Spin Resonance (ESR), indicating that a free radical takes part in this cyclization reaction and acts as an intermediate. Our research is the first report about the application of DESs in supramolecular macrocyclic host synthesis.
Introduction
Deep eutectic solvents (DESs) are composed of mixing halide salts with metal salts or a hydrogen bond donor (HBD).1 DESs can be easily prepared from low cost chemical materials, and through an atom economical approach. In 2001, Abbott2 reported firstly that choline chloride and MCl2 (M = Zn and/or Sn) could form a moisture-stable deep eutectic solvent. In recent decades, DESs have been used widely as Lewis acid catalysts and reaction medium for Diels–Alder reactions,3 O-acetylation of cellulose and monosaccharides,4 Kabachnik–Fields reaction5 and esterification reactions.6Pillar[n]arenes are a new kind of supramolecular macrocyclic host which consist of 1,4-O-disubstituted hydroquinone subunits linked by methylene bridges at their 2- and 5-positions.7 Since they were first discovered by Ogoshi in 2008,8a an increasing number of researches focused not only on their applications in supramolecular chemistry but also on the improvements of their synthesis methods.9 Generally, there are two pathways for the synthesis of pillar[n]arenes, known as the direct method and the indirect method. The direct method is the condensation of 1,4-dialkoxybenzene and paraformaldehyde catalyzed by Lewis acids BF3·OEt2 (ref. 8) and FeCl3 (ref. 10) or organic acid.11 While the indirect method is the cyclooligomerization of 1,4-dialkoxy-2,5-bis(alkoxy-methyl)benzenes in the presence of p-toluenesulfonic acid12 or the cyclization of hydroxyl or bromo substituted 2,5-dialkoxybenzene13 with different kinds of Lewis acids. However, both of the methods have some defects. For example, the yield of pillar[6]arene is poor using the organic acid-catalysed method,11a,c and the highest yield of pillar[6]arene is 45% by using the iron(III) chloride catalysed route.10a Ogoshi has reported the preparation of methyl pillar[5]arene in yields of 71% (ref. 8b) and cyclohexyl pillar[6]arene in yields of 87% (ref. 8c) using BF3·OEt2 as catalyst with the direct method, but the reaction conditions is too harsh. When using the indirect method, the yield of pillar[n]arenes is considerable, but the reaction route is relatively intricate. Nowadays, with the rapid development of pillar[n]arenes in supramolecular chemistry, it is extremely desired to develop a new high-yield synthesis pathway for pillar[n]arenes using a new catalytic systems.
Herein, we initiate a new and distinct method of preparing pillar[5]arene and pillar[6]arene with high selectivity and efficiency by the condensation of 1,4-dialkoxybenzene and paraformaldehyde catalyzed by choline chloride (ChCl)/ferric chloride (FeCl3) DES in CH2Cl2 at room temperature. Under the optimal conditions, the yield of pillar[5]arene and pillar[6]arene can be obtained with the yield of 35% and 53%, respectively. To the best of our knowledge, the research is the first report about the application of DESs in supramolecular macrocyclic host synthesis, which will stimulate the applications of DESs in different relative fields.
Results and discussion
Firstly, the cyclization of 1,4-diethoxybenzene (1a) with paraformaldehyde was chosen as a model reaction (Scheme 1).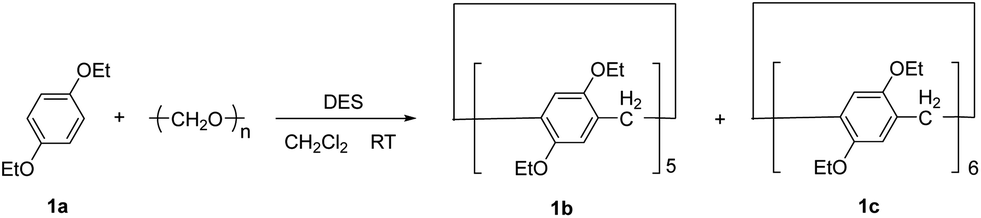 | ||
| Scheme 1 Condensation of 1,4-diethoxybenzene and paraformaldehyde with the deep eutectic solvent. | ||
Then, a series of DESs including [ChCl][MCl]2 (M = Fe, Zn and Sn) and [ChCl][MCl·H2O]2 (M = Mg, Al, Cr, Mn, Co, Ni and Cu) were selected to study their influence on the synthesis reaction of pillar[n]arenes. As shown in Table 1, it was found that the cyclization only occurred in the presence of [ChCl][FeCl3]2 DES (Table 1, entry 1). However, other DESs, such as [ChCl][ZnCl2]2 (Table 1, entry 2–10), had no effect on this reactions even if the reaction time was prolonged to 24 hours. In order to investigate the influence caused by solvents, 1,2-dichloroethane (DCE), CHCl3 and chlorocyclohexane (Cl-CyC6) were selected as reaction mediums to carry out the model reaction. As a result, the yield of 1b in DCE was nearly the same as in CH2Cl2, but the yield of 1c was rather low (Table 1, entry 12). However, when CHCl3 or Cl-CyC6 was utilized as solvent, it only gave poor yields of 1b and 1c (Table 1, entry 13 and 14). Interestingly, this result was totally different from others' works.8c
| Entry | Catalyst | Time | Solvent | Yieldb of 1b | Yieldb of 1c |
|---|---|---|---|---|---|
| a Reaction conditions: 1a (1 mmol), paraformaldehyde (3 mmol), and DESs (0.15 mmol) in CH2Cl2 at room temperature (25 °C).b Isolated yields.c The same reaction conditions as in ref. 10a.d The same reaction conditions as in ref. 8d.e DCE = 1,2-dichloroethane. | |||||
| 1 | [ChCl][FeCl3]2 | 4 h | CH2Cl2 | 35 | 53 |
| 2 | [ChCl][ZnCl2]2 | 4–24 h | CH2Cl2 | 0 | 0 |
| 3 | [ChCl][SnCl2]2 | 4–24 h | CH2Cl2 | 0 | 0 |
| 4 | [ChCl][MgCl2·6H2O]2 | 4–24 h | CH2Cl2 | 0 | 0 |
| 5 | [ChCl][AlCl3·6H2O]2 | 4–24 h | CH2Cl2 | 0 | 0 |
| 6 | [ChCl][CrCl3·6H2O]2 | 4–24 h | CH2Cl2 | 0 | 0 |
| 7 | [ChCl][MnCl2·4H2O]2 | 4–24 h | CH2Cl2 | 0 | 0 |
| 8 | [ChCl][CoCl2·6H2O]2 | 4–24 h | CH2Cl2 | 0 | 0 |
| 9 | [ChCl][NiCl2·6H2O]2 | 4–24 h | CH2Cl2 | 0 | 0 |
| 10 | [ChCl][CuCl2·2H2O]2 | 4–24 h | CH2Cl2 | 0 | 0 |
| 11 | No catalyst | 24 h | CH2Cl2 | 0 | 0 |
| 12 | [ChCl][FeCl3]2 | 4 h | DCEe | 48 | 18 |
| 13 | [ChCl][FeCl3]2 | 4 h | CHCl3 | 2 | Trace |
| 14 | [ChCl][FeCl3]2 | 4 h | Cl-CyC6 | 3 | Trace |
| 15c | FeCl3 | 2 h | CHCl3 | 30 | 34 |
| 16d | BF3·OEt2 | 20 min | CHCl3 | 20 | 15 |
To validate the interesting results shown above, the reactions with FeCl3 based DESs were further investigated. As shown in Table 2, the amount of [ChCl][FeCl3]2 significantly influences the yield of cyclic pentamers. The yield of cyclic pentamers 1b increased with the rasing amount of [ChCl][FeCl3]2 (Table 2, entry 1–6). From 5 mol% to 30 mol% of [ChCl][FeCl3]2, the yield of 1b increased from 22% to 40%. However, with the addition of [ChCl][FeCl3]2 (Table 2, entry 1–6), the yield of 1c increased in a different way from 1b. The yield of 1c reached the summit in the presence of 15 mol% [ChCl][FeCl3]2 (Table 2, entry 3). When the amount of [ChCl][FeCl3]2 further increased from 15 mol% to 30 mol% (Table 2, entry 4–6), the yield of pillar[6]arene declined, instead. Moreover, the influence caused by the formation of DESs was also investigated. When Lewis basic mixtures were formed with iron(III) chloride and choline chloride at the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 or 1:2,14 neither [ChCl]3[FeCl3] (Table 2, entry 7) nor [ChCl]2[FeCl3] (Table 2, entry 8) could promote this reaction. Compared with [ChCl][FeCl3]2, the catalytic efficiency of [ChCl][FeCl3]3 (Table 2, entry 9) was almost the same. Furthermore, when choline chloride in the DES was replaced by acetylcholine chloride, the [AcetylCl][FeCl3]2 DES showed low inefficiency in synthesizing both of 1b and 1c (Table 2, entry 10). In summary, the best practical and the highest efficient method for the synthesis of pillar[n]arenes (n = 5 and 6) is in the presence of 15 mol% [ChCl][FeCl3]2 in CH2Cl2 at room temperature, giving the yield of 1b and 1c 35% and 53%, respectively. The yield of 1c is the highest yield among all the yields reported. It indicates that DES system should be a good choice in the synthesis of pillar[6]arene.
:3 or 1:2,14 neither [ChCl]3[FeCl3] (Table 2, entry 7) nor [ChCl]2[FeCl3] (Table 2, entry 8) could promote this reaction. Compared with [ChCl][FeCl3]2, the catalytic efficiency of [ChCl][FeCl3]3 (Table 2, entry 9) was almost the same. Furthermore, when choline chloride in the DES was replaced by acetylcholine chloride, the [AcetylCl][FeCl3]2 DES showed low inefficiency in synthesizing both of 1b and 1c (Table 2, entry 10). In summary, the best practical and the highest efficient method for the synthesis of pillar[n]arenes (n = 5 and 6) is in the presence of 15 mol% [ChCl][FeCl3]2 in CH2Cl2 at room temperature, giving the yield of 1b and 1c 35% and 53%, respectively. The yield of 1c is the highest yield among all the yields reported. It indicates that DES system should be a good choice in the synthesis of pillar[6]arene.
| Entry | Catalyst (mol%) | Yieldb of 1b | Yieldb of 1c |
|---|---|---|---|
| a Reaction conditions: 1a (1 mmol), paraformaldehyde (3 mmol), and DESs (0.05–0.30 mmol) in CH2Cl2 at room temperature (25 °C).b Isolated yields. | |||
| 1 | [ChCl][FeCl3]2 (5) | 22 | 21 |
| 2 | [ChCl][FeCl3]2 (10) | 26 | 36 |
| 3 | [ChCl][FeCl3]2 (15) | 35 | 53 |
| 4 | [ChCl][FeCl3]2 (20) | 32 | 51 |
| 5 | [ChCl][FeCl3]2 (25) | 38 | 44 |
| 6 | [ChCl][FeCl3]2 (30) | 38 | 43 |
| 7 | [ChCl]3[FeCl3] (15) | 0 | 0 |
| 8 | [ChCl]2[FeCl3] (15) | 0 | 0 |
| 9 | [ChCl][FeCl3]3 (15) | 36 | 48 |
| 10 | [AcetylCl][FeCl3]2 (15) | 24 | 28 |
The alkoxy used in the model reaction was ethyoxyl, and a number of alkoxys with different chain length were also chosen to evaluate the scope of the reaction. The results in Table 3 showed that the cyclization with different alkoxys were all very satisfactory. Noticeably, the overwhelming majority product was 2b by the cyclization of 1,4-dimethoxybenzene (2a) and paraformaldehyde under the optimal conditions. However, the higher cyclooligomer 2c was so poor yielded that it could be only detected by mass spectrometry. Using 1, 4-dibutyloxybenzene (3a) as the starting material, the yield of cyclic pentamers 3b and cyclic hexamers 3c was 33% and 47%, respectively. The yield of them were both a little lower than that of 1b and 1c. As for other starting compounds (R = n-hexyl, n-octyl), it showed that the amount of the two major products reduced and the oligomers were detected by 1H NMR. These results indicates that with the increase of chain length of alkoxy substituents, the rate of the cyclization of 1,4-dimethoxybenzene with paraformaldehyde decelerates, which is in accord with ref. 11a.

Finally, the reaction mechanism was initially investigated by room-temperature X-band Electron Spin Resonance (ESR). The mixture of reactants 1,4-diethoxybenzene (1a) and paraformaldehyde showed no ESR signals. At the moment [ChCl][FeCl3]2 was added, the reaction started. One minute later, two strong ESR signals were observed. These signals represented that free radical and iron(III) species occurred in the reaction system. (Fig. 1, black line). The signal intensity of ESR in the reaction declined as the reaction processed (Fig. 1, red and blue line). After 60 minutes of reaction, the free radical signal disappeared and there was only Fe3+ signal left (Fig. 1, green line). To confirm the role that free radical played in this system, two control experiments were carried out with 1a, paraformaldehyde, [ChCl][FeCl3]2 and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), a free radical capturing reagent. In the first experiment, TEMPO was added at the beginning. As expected, no products could be obtained with the existence of TEMPO. In the second experiment, TEMPO was added to the system after an hour of reaction. As a result, poor yield of pillar[n]arenes were achieved, and the conversion of 1a was low, by contrast (Scheme 2). The ESR results and the experiments reveals that free radical takes part in this cyclization reaction and acts as an intermediate. Further studies will focus on the structure of the free radical, and the pathway of its formation and consumption.
 | ||
| Fig. 1 Room-temperature X-band ESR spectra: during the reaction proceeded between 1 min to 60 min. | ||
 | ||
| Scheme 2 Mechanistic study on the reaction. | ||
Conclusions
In conclusion, a novel method for the effective synthesis of pillar[n]arenes (n = 5 and 6) by condensation of 1,4-dialkoxybenzene and paraformaldehyde catalyzed by [ChCl][FeCl3]2 deep eutectic solvent under mild conditions have been achieved. This method makes it possible to synthesize pillar[6]arene easily and efficiently. The effect of different Lewis acids DESs and the amount of DESs on the cyclization reaction are investigated. The FeCl3 based DESs stands out among all the catalysts reached. Furthermore, different alkoxy substituted pillar[n]arenes (n = 5 and 6) are obtained through this method, and the chain length of alkoxy substituents can obviously influence the reaction rate and yield. The reaction were carried out in accordance with the free radical mechanism. Indeed, the exploration towards the characteristics and utilization of pillar[n]arenes in supramolecular chemistry field would benefit from our findings.Experimental
Materials
All reagents and solvents for syntheses were purchased from commercial sources and used without further purification. It is important to note that the solvents are commercially available without additional drying. 1, 4-dibutyloxybenzene (3a), 1, 4-dihexyloxybenzene (4a) and 1, 4-dioctyloxybenzene (5a) were synthesized according to the paper.15Measurements
The 1H and 13C NMR spectra were recorded on a Bruker 400 MHz NMR spectrometer at 298 K. The chemical shifts (δ) were given in part per million relative to internal tetramethylsilane (TMS, 0 ppm for 1H), CDCl3 (77.3 ppm for 13C). ESI-MS measurement was performed on Thermo Finnigan LCQ advantage at 298 K. All MALDI-TOF-MS spectra were recorded with an Axima TOF2 mass spectrometry. EPR spectra were recorded on a Bruker X-band A200 spectrometer. The solution sample was taken out into a small tube and then analyzed by EPR. EPR spectra was recorded at 298 K on EPR spectrometer operating at 9.420 GHz. Typical spectrometer parameters were: scan range, 3000 G; center field set, 3361 G; time constant, 163.84 ms; scan time, 30.00 s; modulation amplitude 2.0 G; modulation frequency 100 kHz; receiver gain 1.00*104; microwave power, 19.71 mW.Preparation of deep eutectic solvent
A mixture of the ferric chloride (FeCl3) and choline chloride in a molar ratio of 2:1 was heated to 100 °C with gentle stirring until a dark brown clear liquid formed.
General synthetic process of pillar[n]arenes (see ESI† for more details)
Representative synthetic process of pillar[n]arenes: 1b and 1c. To the solution of 1,4-diethoxybenzene (1a) (1.6620 g, 10 mmol) in dichloromethane (150 ml) was added paraformaldehyde (0.9000 g, 30 mmol). And then, [ChCl][FeCl3]2 (0.6970 g, 1.5 mmol) was added to the solution. After the mixture stirred at 25 °C for 4 h, the reaction was quenched by addition of water. The organic phase was separated and washed with saturated aqueous NaHCO3, H2O, and brine. The crude product was purified by column chromatograph to yield 1b (CH2Cl2/petroleum ether = 3:1). 1H NMR (400 MHz, CDCl3): δ 6.73 (s, 1H), 3.83 (q, J = 6.92 Hz, 2H), 3.77 (s, 1H), 1.27 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 149.8, 128.4, 115.0, 63.7, 29.8, 15.1. HR ESI-MS calcd. for C55H70O10Na [M + Na]+ 913.4867, found 913.4. 1c (CH2Cl2/petroleum ether = 100:1). 1H NMR (400 MHz, CDCl3): δ 6.69 (s, 1H), 3.88–3.74 (m, 3H), 1.29 (t, J = 5.9 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 150.4, 127.8, 115.2, 64.0, 30.9, 15.2. HR ESI-MS calcd for C66H84O12Na [M + Na]+ 1091.5860, found 1091.5.
Acknowledgements
This work was financially supported by the National Science Foundation of China (no. 20972120).Notes and references
- Q. Zhang, K. D. Vigier, S. Royer and F. Jerome, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- A. P. Abbott, G. Capper, D. L. Davies, H. Munro, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2001, 2010–2011 RSC.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Green Chem., 2002, 4, 24–26 RSC.
- A. P. Abbott, T. J. Bell, S. Handa and B. Stoddart, Green Chem., 2005, 7, 705–707 RSC.
- S. T. Disale, S. R. Kale, S. S. Kahandal, T. G. Srinivasan and R. V. Jayaram, Tetrahedron Lett., 2012, 53, 2277–2279 CrossRef CAS PubMed.
- Y. Yang, W. He, C. Jia, Y. Ma, X. Zhang and B. Feng, J. Mol. Catal. A: Chem., 2012, 357, 39–43 CrossRef CAS PubMed.
- (a) P. J. Cragg and K. Sharma, Chem. Soc. Rev., 2012, 41, 597–607 RSC; (b) H. Zhang and Y. Zhao, Chem.–Eur. J., 2013, 19, 16862–16879 CrossRef CAS PubMed.
- (a) T. Ogoshi, S. Kanai, S. Fujinami, T. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed; (b) T. Ogoshi, T. Aoki, K. Kitajima, S. Fujinami, T. Yamagishi and Y. Nakamoto, J. Org. Chem., 2011, 76, 328–331 CrossRef CAS PubMed; (c) T. Ogoshi, N. Ueshima, T. Akutsu, D. Yamafuji, T. Furuta, F. Sakakibara and T. i. Yamagishia, Chem. Commun., 2014, 50, 5774–5777 RSC; (d) X. B. Hu, Z. Chen, L. Chen, L. Zhang, J. L. Hou and Z. T. Li, Chem. Commun., 2012, 48, 10999–11001 RSC.
- T. Ogoshi and T. Yamagishi, Chem. Commun., 2014, 50, 4776–4787 RSC.
- (a) H. Tao, D. Cao, L. Liu, Y. Kou, L. Wang and H. Meier, Sci. China: Chem., 2012, 55, 223–228 CrossRef CAS; (b) L. Liu, D. Cao, Y. Jin, H. Tao, Y. Kou and H. Meier, Org. Biomol. Chem., 2011, 9, 7007–7010 RSC.
- (a) K. Wang, L. L. Tan, D. X. Chen, N. Song, G. Xi, S. X. A. Zhang, C. Li and Y. W. Yang, Org. Biomol. Chem., 2012, 10, 9405–9409 RSC; (b) T. Boinski and A. Szumna, Tetrahedron, 2012, 68, 9419–9422 CrossRef CAS PubMed; (c) C. Han, F. Ma, Z. Zhang, B. Xia, Y. Yu and F. Huang, Org. Lett., 2010, 12, 4360–4363 CrossRef CAS PubMed.
- D. Cao, Y. Kou, J. Liang, Z. Chen, L. Wang and H. Meier, Angew. Chem., Int. Ed., 2009, 48, 9721–9723 CrossRef CAS PubMed.
- (a) Y. Ma, Z. Zhang, X. Ji, C. Han, J. He, Z. Abliz, W. Chen and F. Huang, Eur. J. Org. Chem., 2011, 5331–5335 CrossRef CAS; (b) M. Holler, N. Allenbach, J. Sonet and J. F. Nierengarten, Chem. Commun., 2012, 48, 2576–2578 RSC.
- A. P. Abbott, G. Capper, D. L. Davies and R. Rasheed, Inorg. Chem., 2004, 43, 3447–3452 CrossRef CAS PubMed.
- P. Paduraru, R. Popoff, R. Nair, R. Gries, G. Gries and E. Plettner, J. Comb. Chem., 2008, 10, 123–134 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, preparation of deep eutectic solvents, synthesis process of pillar[n]arenes and characterization data of all compounds. See DOI: 10.1039/c4ra15758c |
| This journal is © The Royal Society of Chemistry 2015 |