
Panoptically exfoliated morphology of chlorinated polyethylene (CPE)/ethylene methacrylate copolymer (EMA)/layered silicate nanocomposites by novel in situ covalent modification using poly(ε-caprolactone)†
Abstract
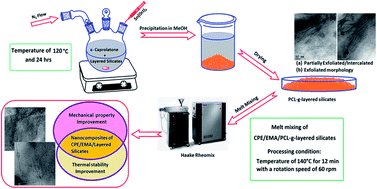
Nanocomposites of a chlorinated polyethylene (CPE)/ethylene methacrylate copolymer (EMA)/layered silicate were prepared by a direct melt blending technique and masterbatch process using two varieties of layered nanosilicates namely; polar Cloisite 30B with two hydroxyl groups in its organic modifier and non-polar Cloisite 20A. The first step was the synthesis of poly(ε-caprolactone) (PCL) grafted layered silicate masterbatch by covalent modification of in situ ring opening polymerization (ROP) of ε-caprolactone and subsequent characterization, followed by melt mixing of this layered silicate masterbatch into a CPE/EMA blend with a 60/40 ratio. A morphology study using wide angle X-ray diffraction (WXRD) and transmission electron microscopy (TEM) indicates complete exfoliation in the masterbatch PCL-g-30B and its nanocomposite (C60E40/PCL-g-30B). The addition of PCL-g-layered nanosilicate masterbatches with a higher degree of exfoliation, promoted the polymer–filler interaction as evident from Fourier transform infrared (FTIR) analysis which improved the static and dynamic mechanical properties and glass transition temperatures (Tg) of the nanocomposites. Fractography from scanning electron microscopy (SEM) images and topographical analysis using atomic force microscopy (AFM) clearly depicted improved filler dispersion in the PCL masterbatch based nanocomposites which also showed improved thermal stability to the CPE/EMA blend.
 Please wait while we load your content...
Please wait while we load your content...

