
Molecular insight into the mode-of-action of phosphonate monolayers as active functions of hybrid metal oxide adsorbents. Case study in sequestration of rare earth elements†
Abstract
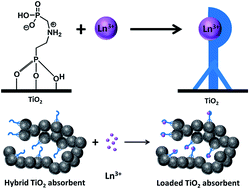
The insight into the molecular aspects of ligand grafting and potential maximal capacity of hybrid organic–inorganic adsorbents bearing phosphonate ligand monolayers as active functions was obtained by single crystal X-ray studies of ligand-functionalized titanium alkoxide complexes. The attachment of molecules occurs generally in the tripodal vertical fashion with the minimal distance between them being about 8.7 Å, resulting in 0.19 nm2 as the minimal surface area per function. In the present experimental work the theoretical loading capacity could almost be achieved for functionalization of mesoporous nanorods of anatase with imino-bis-methylphosphonic acid (IMPA, NH(CH2PO3H2)2) or aminoethylphosphonic acid (AEPA, H2NC2H4PO3H2). The products had the same morphology as the starting material, as was established by SEM and optical microscopy. The size and structure of the individual nanoparticles of the constituting inorganic component of the material were preserved and practically unchanged through the surface modification, as established by powder XRD and EXAFS studies. The surface area of the inorganic–organic hybrids decreased somewhat from the initial ∼250 m2 g−1, on adsorption of AEPA (0.21 mmol g−1) to ∼240 m2 g−1, and on adsorption of IMPA (0.17 mmol g−1) to ∼190 m2 g−1. The ligands were bound effectively to the surface according to TGA, EDS and FTIR analyses and remained in the mono-deprotonated form. The produced hybrid adsorbents had for the selected pH (3.5) high capacities towards adsorption of Rare Earth Element (REE) cations, but with equilibria achieved relatively slowly. The composition of the surface complexes was determined as M : L = 1 : 1 for IMPA, but varied for the AEPA from 1 : 3 to 1 : 1 dependent on the REE, which can be interpreted in terms of charge compensation as the major driving force behind binding. The cation desorption in strongly acidic media for recuperation of the adsorbed REE and the relative capacity of the re-used adsorbent have been quantified.
 Please wait while we load your content...
Please wait while we load your content...

