
In silico de novo design of novel NNRTIs: a bio-molecular modelling approach†
Abstract
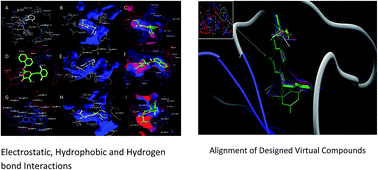
Six novel non-nucleoside reverse transcriptase inhibitors exhibiting high efficacy are designed using in silico mathematical modelling techniques and the results are validated using a docking technique. An in silico assessment of interaction potential and structural requirements of 5-alkyl-2-alkylamino-6-(2,6-difluorophenylalkyl)-3,4-dihydropyrimidin-4(3H)-one (DABO) analogues in the non-nucleoside inhibitor binding pocket is also performed. Efficient use of 3D-pharamacophoric (SALL, HDALL, HAALL and RALL) and 3D-averaged alignment (C log P and dipole moment) descriptors is made in this study. The chemometric analyses, using support vector machine, back propagation neural network and multiple linear regression, are performed. The relative potentials of these chemometric methods is also assessed and the results, SVM (r = 0.939, MSE = 0.071, q2 = 0.876), BPNN (r = 0.923, MSE = 0.104, q2 = 0.818) and MLR (r = 0.912, MSE = 0.096, q2 = 0.832), indicates that SVM describes the relationship between the descriptors and inhibitory activity in a better manner. The results also suggest that there is a non-linear relationship between the descriptors and inhibitory activity. The study further suggests that isopropyl/propenyl groups as R and R′, oxobutyl group as X and di or tri-substitution as R′′ are the best suited substituents for exhibiting better inhibitory activity.
 Please wait while we load your content...
Please wait while we load your content...

