
Light-driven reversible strand displacement using glycerol azobenzene inserted DNA†
Abstract
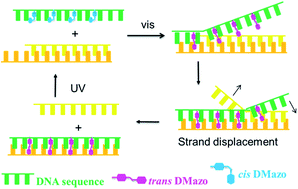
Tethered with bistable 2′,6′-dimethylazobenzene (DMazo) via a glycerol linker, an artificial 35 nt-long DNA has performed photoresponsive hybridization and reversible light-driven strand displacement.
 Please wait while we load your content...
Please wait while we load your content...

