
Benzimidazolin-2-ylidene N-heterocyclic carbene complexes of ruthenium as a simple catalyst for the N-alkylation of amines using alcohols and diols†
Siah Pei Shana,
Xie Xiaokeb,
Boopathy Gnanaprakasam‡
a,
Tuan Thanh Danga,
Balamurugan Ramalingama,
Han Vinh Huynh*b and
Abdul Majeed Seayad*a
aInstitute of Chemical and Engineering Sciences, 8 Biomedical Grove, #07-01 Neuros, 138665, Singapore. E-mail: abdul_seayad@ices.a-star.edu.sg
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore. E-mail: chmhhv@nus.edu.sg
First published on 9th December 2014
Abstract
Simple air and moisture stable ruthenium complexes 1–3 and 3a were synthesized from readily available benzannulated N-heterocyclic carbene ligands (bimy = benzimidazolin-2-ylidene). These complexes were found to be efficient catalysts for the alkylation of amines using alcohols as alkylating agents. Catalysts 1, 2 and 3a gave excellent yields of up to 99% for the alkylation of various amines using benzylic and aliphatic alcohols at 130 °C for 18 h under solventless conditions. Catalyst 3a bearing both phosphine and carbene ligands gave excellent yields of up to 98% for the synthesis of heterocyclic amines by double alkylation of primary amines using linear diols. The practical utility of these catalysts was demonstrated for the synthesis of pharmaceutically important amines in a more environmentally benign way under solventless conditions.
Introduction
Molecules that contain C–N bonds are key intermediates in the synthesis of bioactive molecules and play an important role in the pharmaceutical industry.1 Traditional methods for C–N bond formation including SN2 type amination using toxic alkyl halides2 and reductive amination using stoichiometric amounts of reducing agents3 are not environmentally benign or atom economical.4 Recently, transition metal catalyzed C–N bond formation from alcohols and amines following the so-called “hydrogen borrowing” methodology,5 has attracted much attention. Alcohols are often low-cost, less hazardous and commercially available starting materials, and water is the only by-product of this reaction, which makes this method environmentally attractive.6 Pioneering works in this field have been done by the groups of Grigg7 and Watanabe.8 Great contributions have also been made by the groups of Crabtree,9 Williams,10 Milstein,11 Beller12 and Fujita.13 In most of the cases, ruthenium14 or iridium15 complexes have been used as catalysts to achieve efficient N-alkylation of a variety of amines or ammonia with primary or secondary alcohols including diols. In recent years, catalysts based on other noble metals such as Au,16 Ag,17 Os,18 Rh,19 and Pd20 as well as non-noble metals such as Fe,21 Cu,22 Ni,23 Bi,24 In25 and Re26 were also reported to be effective as catalysts for the use of alcohols as N-alkylating agents adopting hydrogen borrowing strategy.N-heterocyclic carbenes (NHCs) have become ubiquitous ligands for homogeneous catalysis,27 and NHC complexes of ruthenium and iridium have been reported to be active catalysts for the activation of alcohols by hydrogen borrowing strategy. As an early example, Cp*-functionalized iridium–carbene complexes have been reported28 by Peris et al. for the alkylation of aniline and β-alkylation of secondary alcohols. Up to 87% of yield was observed in the presence of strong bases such as tBuOK (110 mol%) at 110 °C. Subsequently, they also reported29 the application of imidazolin-2-ylidene, imidazolin-4-ylidene and pyrazolin-3-ylidene derived ruthenium complexes as catalyst for the β-alkylation of secondary and primary alcohols. In the presence of one equivalent of KOH, quantitative yield of the β-alkylated alcohols were formed in toluene at 110 °C. Crabtree et al.9a reported chelating pyrimidine–NHC complexes of iridium and ruthenium as effective catalysts for the alkylation of amines and secondary alcohols. Using these catalysts, N-alkylation of both electron deficient and electron rich amines was carried out in the presence of NaHCO3 (50 mol%) to give the corresponding secondary amines in 25–98% yield. Recently, Valerga et al.30a reported the N-alkylation of both aromatic and non-aromatic amines using Ru(II)-picolyl–NHC complex30b as a catalyst. Turn over numbers (TON) of up to 480 were reported in the presence of 50 mol% of KOH at 100 °C.
Bifunctional Ir catalysts bearing alcohol/alkoxide tethered NHC have been reported for N-alkylation of various amines at temperature as low as 50 °C.15c The catalysts were reported to have the ability to accept both proton and the hydride in order to form the products in 77–99% yield in the absence of any external base. Anderson et al. reported Ir complexes of chelating ligand containing phosphine and NHC moiety as catalysts for N-monoalkylation of amines.15m Notably, the catalyst promoted the alkylation at 50 °C and at room temperature for selected substrates, in the presence of 50 mol% of tBuOK to give the alkylated products in 63–97% yields.
To the best of our knowledge, only imidazolin-2-ylidene29 based complexes having chelating N-pyrimidyl9a or N-picolyl30 substituents have been reported for the N-alkylation of amines, while simpler non-chelating or benzimidazolin-2-ylidene based ruthenium complexes have not been explored yet. Herein, we report simple and non-chelating Ru(II)–NHC complexes of the general formula [RuCl2(p-cymene)(bimy)] (bimy = benzimidazolin-2-ylidene) as efficient catalysts for the N-alkylation of amines using alcohols.
Results and discussion
The preparation of complexes [RuCl2(p-cymene)(iPr2-bimy)] (1) and [RuCl2(p-cymene)(Bn2-bimy)] (3) bearing symmetrical benzimidazolin-2-ylidene ligands were reported previously.31 Their preparation is improved in this work, and in analogy, the new complex [RuCl2(p-cymene)(iPr, Bn-bimy)] (2) containing an unsymmetrically substituted NHC was readily synthesized by treating the [RuCl2(p-cymene)]2 dimer directly with the corresponding Ag-carbene species, that in turn was generated in situ by mixing the known benzimidazolium salt B32 with Ag2O in CH2Cl2 (Scheme 1).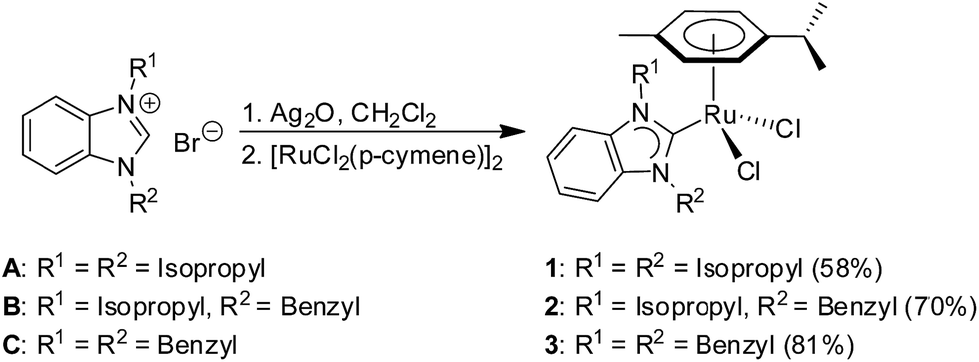 | ||
| Scheme 1 Synthesis of ruthenium NHC complexes 1–3. | ||
In terms of stereoelectronic properties, complex 2 represents a midway between complexes 1 and 3.32 Pure samples can be obtained by washing the crude products with water and diethyl ether. The yields of complexes 1, 2 and 3 were 58%, 70% and 81%, respectively. Notably, the reaction for the preparation of complex 1 is more sluggish than that for complex 2 and 3. About 20% of unreacted Ru dimer can still be observed by 1H NMR analysis after 12 h. The lower acidity of the C2–H proton in precursor A as a consequence of the positive inductive effects from two N-isopropyl substituents makes the generation of the required silver carbene species comparatively more challenging. Furthermore, the bulkier isopropyl groups in precursor A could also slow down the carbene transfer rate from the Ag complex to the Ru dimer.
Complexes 1–3 are soluble in most organic solvents, such as CH2Cl2, CHCl3, MeOH and CH3CN, with the exception of nonpolar ones, such as hexane and diethyl ether. Their formation is supported by ESI mass spectra, where base peaks corresponding to the [M − 2Cl + OH + MeCN]+ fragment were observed. In the 1H NMR spectra, the absence of downfield signals characteristic for the benzimidazolium salts indicates the formation of the expected Ru–NHC complexes. The carbene signals in the 13C NMR spectra of complexes 1–3 are observed at 187.4, 189.5 and 191.6 ppm, respectively, which are more downfield compared to their imidazolin-2-ylidene analogues (ca. 170 ppm).33 The gradual downfield shifts of the 13Ccarbene signals in these three complexes is reflective of the decreasing +I effect of the wing tip N-substituents on the carbene ligands.
The molecular structure of 2 (Fig. 1) was determined by X-ray diffraction on single crystals obtained by diffusion of diethyl ether into a dichloromethane solution. This complex adopts three-legged piano stool geometry with the facial planar p-cymene representing the “seat”, and the two chlorido and one NHC ligands form the three “legs”. Although the carbon atom C21 is situated “transoid” to the carbene donor C1 (C21–Ru1–C1 = 162.13°), no trans influence from the NHC ligand can be discerned as all Ru–Ccymene bonds are of essentially the same length within 3σ with an average value of 2.209 Å. The Ru-centroid distance amounts to 1.693 Å. Both the N-benzyl group and the N-isopropyl group are pointing away from the ruthenium center to avoid steric repulsion, and the bulkier isopropyl group in the p-cymene ring is also oriented away from the benzimidazolin-2-ylidene ligand for the same reason.
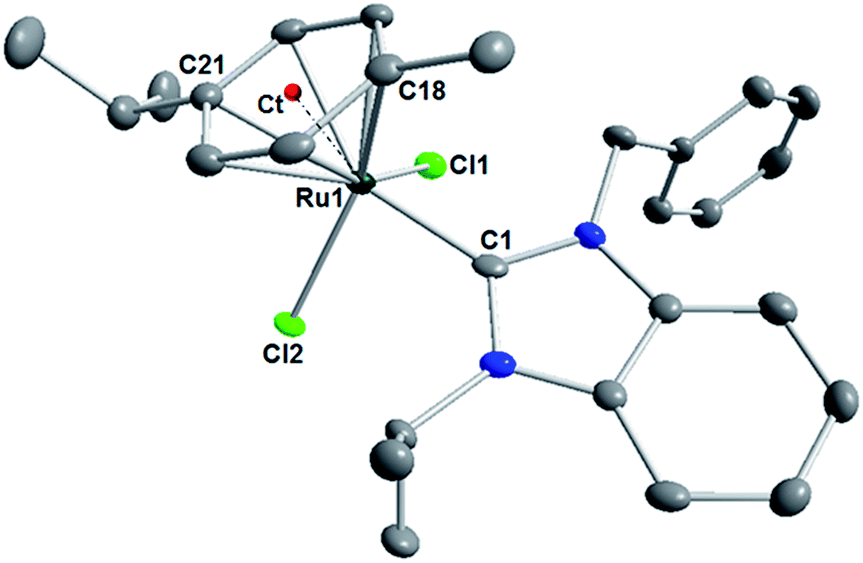 | ||
| Fig. 1 Molecular structure of complex 2 showing 50% probability ellipsoids. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and bond angles (deg): Ru1–C1 2.0692(24), Ru1–Cl1 2.4096(10), Ru1–Cl2 2.4381(9); Ru1–Cl18 2.2073(24), Ct–Ru1 1.693; Ru1–Cl21 2.2638(26); Cl1–Ru1–Cl2 83.795(2), Cl1–Ru1–C1 89.200(6), Cl2–Ru1–C1 91.721(7). | ||
The catalytic activity of the ruthenium benzimidazolin-2-ylidene complexes 1–3 for alcohol activation towards N-alkylation was investigated by taking the coupling between benzyl alcohol and aniline as a standard reaction (Table 1). Catalyst 1 having the NHC ligand symmetrically substituted with isopropyl group was selected for initial optimization of the reaction conditions. Only a moderate 40% yield (entry 1) of the N-benzylamine product was obtained after 18 h at a lower catalyst loading of 1.0 mol% and at a reaction temperature of 130 °C under solventless conditions. The yield increased to 70% upon increasing the amount of the catalyst to 2.5 mol% (entry 2). Increase in temperature showed no significant improvement (entry 3), but the yield decreased when the temperature was lowered (entry 4).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Amount of catalyst (mol%) | Temp. (°C) | Time (h) | Yield, (%) |
| a Reaction conditions: 1 mmol of aniline, 1.2 mmol of benzyl alcohol. % yields were determined by GC analysis using docecane as the internal standard.b Toluene as solvent.c Diglyme as a solvent. | |||||
| 1 | 1 | 1.0 | 130 | 18 | 40 |
| 2 | 1 | 2.5 | 130 | 18 | 70 |
| 3 | 1 | 2.5 | 140 | 18 | 68 |
| 4 | 1 | 2.5 | 120 | 18 | 53 |
| 5 | 1 | 2.5 | 130 | 6 | 60 |
| 6 | 1 | 5.0 | 130 | 6 | 80 |
| 7 | 1 | 5.0 | 130 | 6 | 13b |
| 8 | 1 | 5.0 | 130 | 6 | 8c |
| 9 | 2 | 5.0 | 130 | 6 | 62 |
| 10 | 3 | 5.0 | 130 | 6 | 5 |
| 11 | 1 | 5.0 | 130 | 18 | >99 |
| 12 | 2 | 5.0 | 130 | 18 | >99 |
In general, the initial catalytic activity observed was higher and up to 60% yield was obtained for a 6 h reaction at 130 °C (entry 5). Under these conditions, doubling the catalyst loading improved the yield to 80% (entry 6). At this point, the catalytic performance in presence of a solvent was investigated. The catalytic performance decreased drastically, and only 13% and 8% yields were observed with common solvents generally used for this reaction such as toluene and diglyme (entries 7 & 8), respectively.
Under the present optimized conditions, catalyst 2 having one of the isopropyl substituents replaced with a benzyl group gave a yield of 62% (entry 9). However, the catalytic activity was nearly suppressed when both the isopropyl groups were replaced with benzyl groups as in catalyst 3. The unfavorable effect of the benzyl groups on the catalytic performance may be due to a possible π-coordination34 of benzyl moiety to the Ru-center upon removal of p-cymene or other ligands under the reaction conditions, thus hindering the coordination and further activation of substrates and intermediates. Although catalyst 2 shows slightly lower initial activity compared to 1, both the catalysts (1 and 2) gave near quantitative yields upon increasing the reaction time to 18 h (entries 11 & 12).
The catalytic performances of the benzimidazolin-2-ylidene ruthenium complexes 1–3, were compared with those of some common ruthenium imidazolin- and imidazolidin-2-ylidene complexes (4–7) under the same optimized conditions, and the results are given in Table 2. Complexes 4 and 5 with IMes and IiPr ligands gave lower yields of 24% and 19% (entries 1 & 2), respectively. Grubb's second generation catalyst (6) bearing the SIMes gave an improved yield of 88% while, the Hoveyda-Grubb's catalyst (7) gave up to 74% yield (entries 3 & 4). We reckoned that the improved catalytic performance observed with Grubbs II catalyst 6 compared to other catalysts may be due to the presence of a phosphine (PCy3) as an additional ligand. Hence, the addition of phosphine was investigated to examine if any beneficial synergistic effect exists. No specific effect was observed with the Hoveyda-Grubb's catalyst (7) in the presence of PPh3.35 However, enhanced catalytic activity was noted with catalysts 4 and 5, and up to 51% and 45% yields were observed at 130 °C after 6 h (entries 5 & 6).
|
|||
|---|---|---|---|
| Entry | Catalyst | Additive | Yield (%) |
| a Reaction conditions: 1 mmol of aniline, 1.2 mmol of benzyl alcohol. 5 mol% of Ru–NHC catalyst at 130 °C for 6 h. % yields were determined by GC analysis using docecane as the internal standard. | |||
| 1 | 4 | Nil | 24 |
| 2 | 5 | Nil | 19 |
| 3 | 6 | Nil | 88 |
| 4 | 7 | Nil | 74 |
| 5 | 7 | PPh3 (5 mol%) | 75 |
| 6 | 4 | PPh3 (5 mol%) | 51 |
| 7 | 5 | PPh3 (5 mol%) | 48 |
| 8 | 1 | PPh3 (5 mol%) | 87 |
| 9 | 2 | PPh3 (5 mol%) | 64 |
| 10 | 3 | PPh3 (5 mol%) | 85 |
The performance of catalysts 1 and 2 marginally improved in the presence of PPh3 giving up to 87% and 64% yields, respectively (entries 8 and 9). However, the 1,3-dibenzyl-benzimidazolin-2-ylidene complex 3 became much more active in the presence of PPh3 yielding up to 85% of the product (entry 10). This favorable effect of the phosphine could be due to its coordination to the ruthenium center, which may sterically prevent the π-coordination of benzyl moiety to the Ru-center.
In order to understand this beneficial effect on relatively inactive complexes such as 3, the new mixed NHC–phosphine ruthenium complex [RuCl2(p-cymene)(bimy)Cl(PPh3)]PF6 (3a) was prepared as shown in Scheme 2 for the purpose of further comparison. A small improvement in catalytic activity was observed (87%, under the reaction conditions given in Table 2) with well-defined 3a compared to that of the in situ generated species by external addition of PPh3 to catalyst 3 (85%, Table 2, entry 10). Notably, near quantitative yield was observed for 3a upon increasing the reaction time to 18 h. Since the benzimidazolin-2-ylidene(bimy) complexes 1, 2 and 3a gave similar results for the N-alkylation using primary alcohols under the optimized conditions (at 130 °C for 18 h under solventless conditions), they were further evaluated for substrate scope and limitations.
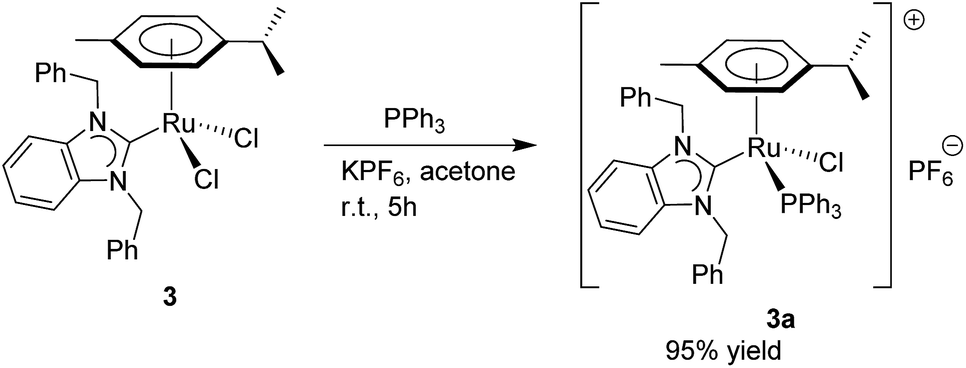 | ||
| Scheme 2 Synthesis of Ru–NHC–phosphine complex 3a. | ||
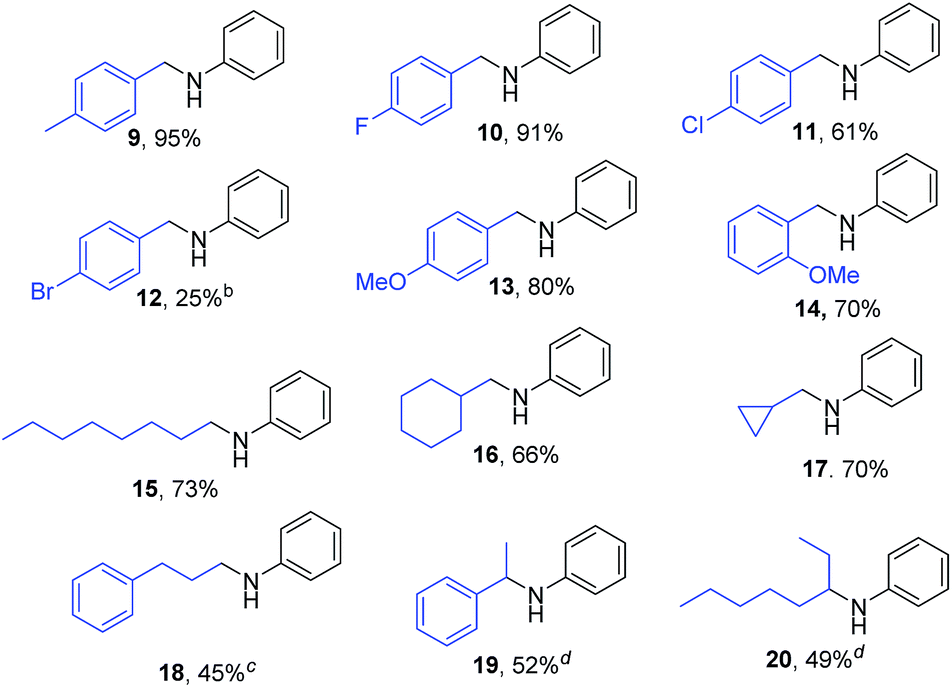
Alkylation of aniline using various benzylic and aliphatic alcohols gave good to excellent results using catalyst 1 as presented in Table 3. Excellent isolated yield of 95% was achieved for the secondary amine 9 when p-Me substituted benzylalcohol was used as the alkylating agent. p-F and p-Cl substituted benzyl alcohols gave the corresponding secondary amine products 10 and 11 in 91% and 61% isolated yields respectively. The p-Br substituted benzyl alcohol showed poor activity yielding 12 in 25% with lower conversion. Both p-OMe and o-OMe substituted benzyl alcohols were coupled with aniline giving the corresponding secondary amine products 13 (80%) and 14 (70%) in very good yields. The relatively less reactive aliphatic alcohols were also found to be promising alkylating agents and up to 73% isolated yield of N-octyl aniline (15) was obtained when 1-octanol was used. Secondary amines such as 16 and 17 were obtained in good yields of 66% and 77% respectively, when cyclohexylmethanol and cyclopropylmethanol were used as the alkylating agents. Aliphatic alcohol such as 3-phenylpropanol gave only moderate yield of 45% of the secondary amine 18 under the present set of conditions. Secondary alcohols such as 1-phenylethanol and 2-octanol were found to be poor alkylating agents in the presence of catalyst 1, 2 and 3a providing maximum yields of 52% (19) and 49% (20).
|
|---|
| a Reaction conditions: 1 mmol of aniline, 1.2 mmol of alcohol, 5 mol% of catalyst 2 at 130 °C for 18 h. Isolated yields are given.b 2 was used as the catalyst; catalysts 1 and 3a gave 18% and 22% yields respectively.c 2 was used as the catalyst; catalysts 1 and 3a gave 40% and 43% yields respectively.d Catalyst 3a was used, catalysts 1 and 2 gave lower yields. |
 |
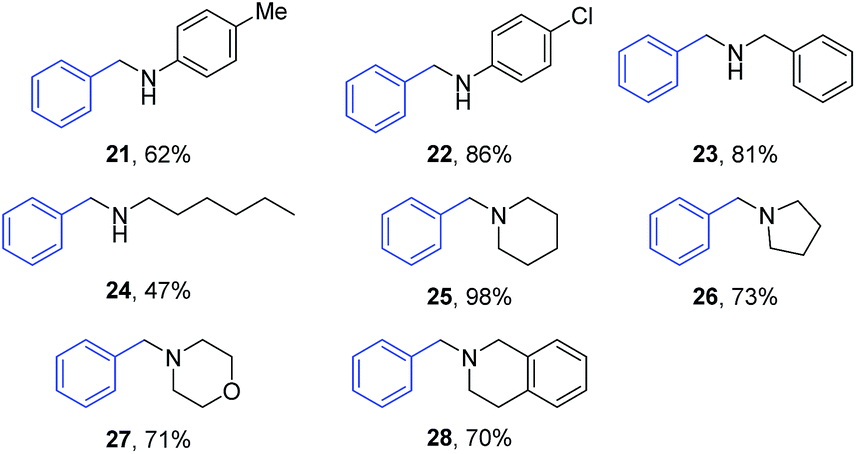
Next, the substrate scope of amines was investigated using benzyl alcohol as the alkylating agent (Table 4). Electron donating p-Me substituted aniline gave 21 in moderate yield of 62%, while an increased yield up to 86% was observed for 22 when p-chloro aniline was used as the substrate. Benzylamine gave dibenzylamine (23) in 81% yield, whereas 47% yield of the secondary amine 24 was observed when an aliphatic amine such as hexylamine was used. Near quantitative yield of the tertiary amine 25 was achieved for the alkylation of six membered cyclic amine such as piperidine. Other cyclic amines such as pyrrolidine, morpholine and 1,2,3,4-tetrahydroisoquinoline were also readily alkylated giving the corresponding cyclic tertiary amines 26–28 in 70–73% isolated yields.
|
|---|
| a Reaction conditions: 1 mmol of amine, 1.2 mmol of benzyl alcohol, 5 mol% of catalyst 1 at 130 °C for 18 h. Isolated yields are given. |
 |

The possible application of the benzimidazolin-2-ylidene complexes 1, 2 and 3a were further examined for the synthesis of pharmaceutically important secondary and tertiary amines. For this purpose, fendiline (29), an anti-anginal agent was chosen as an initial example (Scheme 3). The direct synthesis of fendiline36 was reported previously by Williams and coworkers adopting hydrogen borrowing strategy in presence of [Cp*IrCl2]2 as the catalyst.37
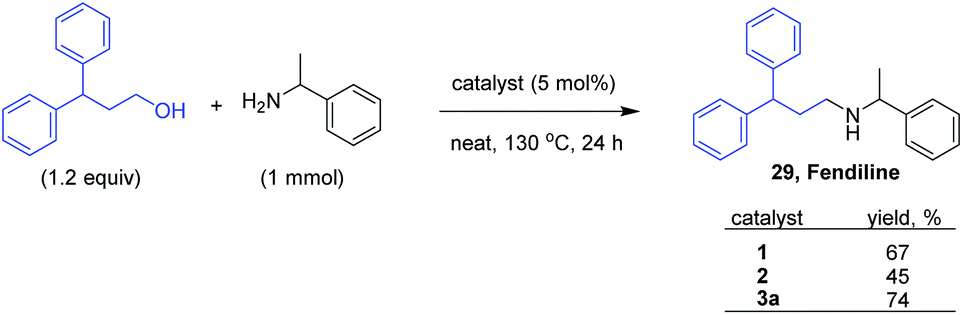 | ||
| Scheme 3 Catalyst optimization for the synthesis of fendiline. | ||
Catalyst 1 having symmetrically substituted isopropyl groups gave up to 69% yield, while the unsymmetrically substituted catalyst 2 gave a lower yield of 43% at 130 °C after 24 h reaction. Interestingly, catalyst 3a gave an improved yield of 74% and was selected for further applications in the synthesis of pharmaceutically important amines. Under the standardized conditions as shown in Scheme 3, good isolated yield of 62–70% were achieved for the synthesis of some of the pharmaceutically important compounds such as alverine38 (30), fenpiprane39 (31) and prozapine39 (32) (Fig. 2). To the best of our knowledge, H-borrowing strategy has not been demonstrated for the synthesis of pharmaceutically important molecules 30–32.
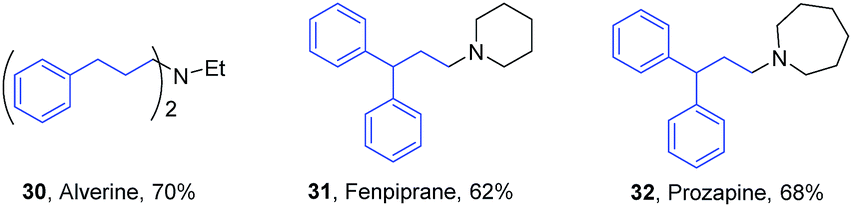 | ||
| Fig. 2 Synthetic application of catalyst 3a for the preparation of pharmaceutically important amines. | ||
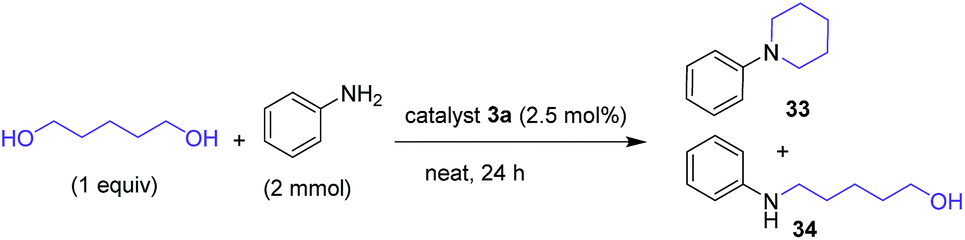
The cyclization using diols following hydrogen borrowing strategy is a convenient protocol to prepare several value-added cyclic amines from the corresponding primary amines. Yamaguchi et al. employed iridium catalyst for the synthesis of cyclic tertiary amines by reaction of various primary amines with suitable diols to achieve good yields.40 Ruthenium catalysts were reported to be effective for using diols as alkylating agents for the preparation of cyclic amines.10a,41 Considering the importance of cyclic amines, we have investigated the catalytic performance of the benzannulated NHC complexes 1, 2 and 3a for their synthesis using diols as alkylating agents.
As shown in Table 5, yields of 23–33% were observed for the cyclized product N-phenyl piperidine (33) with a conversion of 89% at 130 °C after 24 h using 2.5 mol% catalysts 1 and 2. In both the cases, the monoalkylated product 34 was found to be the major product (entries 1 & 2). However, up to 66% yield to 33 was observed with the catalyst 3a, which was further improved to 93% (entry 4) upon increasing the temperature to 150 °C. No specific temperature effect was observed for the catalyst 1 or 2. This optimized condition was used to extend the substrate scope using various diols as alkylating agents for the synthesis of cyclic amines.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Temp. (°C) | Conv. (%) | Yield of 33 (%) | Yield of 34 (%) |
| a Reaction conditions: 2 mmol of aniline, 2 mmol of 1,5-pentanediol. 2.5 mol% of Ru–NHC catalyst for 24 h. Yields were determined by GC analysis using dodecane as the internal standard.b 89% isolated yield. | |||||
| 1 | 1 | 130 | 89 | 33 | 55 |
| 2 | 2 | 130 | 82 | 23 | 58 |
| 3 | 3a | 130 | 96 | 66 | 29 |
| 4 | 3a | 150 | 98 | 93b | 4 |
| 5 | 1 | 150 | 87 | 47 | 38 |
| 6 | 2 | 150 | 86 | 45 | 39 |
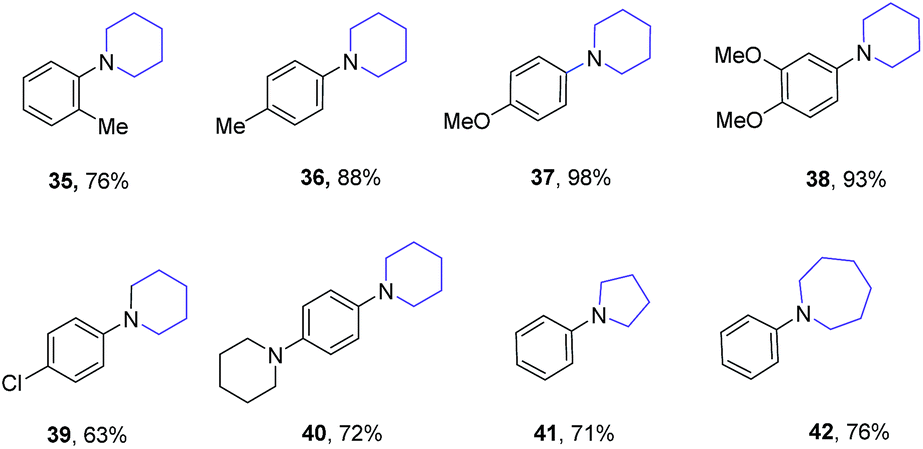
Excellent yields up to 98% were achieved for various anilines as substrates at 150 °C as given in Table 6. Both o-Me and p-Me substituted anilines were double alkylated with 1,5-pentandiol giving the cyclized product 35 and 36 in 76% and 88% yields, respectively. Excellent yields were also achieved for the synthesis of 37 (98%) and 38 (93%) using 4-methoxy and 3,4-dimethoxy substituted anilines. 1-(4-chlorophenyl)piperidine (39) was obtained in 63% yield from 4-chlorophenyl aniline and 1,5-pentandiol. 1,4-di(piperidin-1-yl)benzene (40) was obtained in 73% yield from 4-(piperidin-1-yl)aniline and 1,5-pentanediol. Both 1,4-butanediol and 1,6-hexandiol react smoothly with aniline to form 1-phenylpyrrolidine (41) and 1-phenylazepane (42), respectively, in very good yields.
|
|---|
| a Reaction conditions: 2 mmol of aniline, 2 mmol of diol, 150 °C, 24 h. 2.5 mol% of Ru–NHC catalyst for 24 h. Isolated yields are reported. |
 |
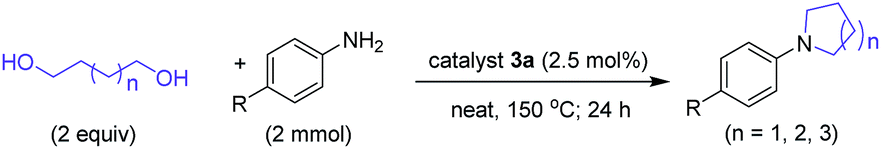
Ruthenium catalyzed alcohol activation is reported to proceed through the formation of a Ru–H species as an active intermediate that initiate the catalytic cycle.42 In the present Ru–NHC catalyst system, formation of a Ru–H species43 at δ = −9.62 ppm was detected by 1H NMR spectroscopy (Fig. 3, Spectrum A) from an intermediate reaction sample after 2 h of the standard N-alkylation reaction44 (Table 1) at 100 °C using catalyst 1. Apart from this signal, additional signals at −1.69 and −2.93 ppm were also observed. In the presence of one additional equivalent of PPh3,44 two doublets at −7.36 (J = 53.4 Hz) and −9.41 ppm (J = 56.2 Hz) as well as two triplets at −9.81 (J = 35.8 Hz) and −13.28 ppm (J = 19.9 Hz) were observed45 showing the formation multiple Ru–H species coordinated with PPh3 (Fig. 3, Spectrum B). The nearly inactive complex 3 showed one prominent Ru–H signal at −8.3 ppm along with a slightly upfield shifted, low intensity broad resonance (Fig. 3, Spectrum C), which may be attributed to a possible formation of stable π-benzyl coordinated species that may restrict coordination of the intermediate imine for completing the catalytic cycle. However, in the presence of added one equivalent of PPh3, this Ru–H species was considerably reduced with the formation of three sets of doublets (δ = −9.09, −9.82 and −10.01; approx. J = 48 Hz) and two sets of triplets (δ = −9.81 and −10.88; J = 35.7 Hz) (Fig. 3, Spectrum D). Some of these hydride signals were found to disappear with time.
 | ||
| Fig. 3 1H NMR spectroscopic (400 MHz) analysis for Ru–H species formed under reaction conditions with complex 1 (A, CDCl3), complex 1 + PPh3 (B, CDCl3), complex 3 (C, CDCl3), complex 3 + PPh3 (D, CDCl3) and complex 3a (E, DMSO-d6).44 | ||
Unfortunately, the intermediate complexes obtained from 3a were not very stable and immediately precipitated as the parent compound 3a upon dissolving in common deuterated solvents (CDCl3, CD3OD, etc.). However, from a dilute solution obtained by warming the reaction mixture in DMSO-d6, we could observe multiple Ru–H signals (Fig. 3, Spectrum E) such as three doublets {−7.61 ppm, (J = 47.3 Hz), −8.01 ppm (d, J = 52.1 Hz) and −10.28 ppm (d, J = 48.3 Hz)}, one triplet at −10.28 ppm (d, J = 48.3 Hz); and one doublet of doublet at −9.18 ppm (J = 49.2, 13.4 Hz). In all these cases two to four sets of p-cymene signals44 were also detected showing the presence of coordinated p-cymene ligand in these intermediate complexes.
In an effort to further characterize the intermediates, 31P{1H}NMR analysis (Fig. 4) was carried out with the samples obtained under reaction conditions from complexes 1 + PPh3 (Spectrum F), 3 + PPh3 (Spectrum G) and 3a (Spectrum H). In the case of 3a, two new species were detected at 27.18 and 25.39 ppm together with free PPh3 (−5.29 ppm) and 3a (33.17 ppm). Complex 3 in the presence of added PPh3 gave a major signal at 24.52 ppm and several 31P NMR signals in the range of 20 to 55 ppm, which were difficult to assign to any particular intermediate species. However, complex 1 with added PPh3 gave five sharp signals in the range of 25 to 55 ppm with a major signal at 24.56 ppm. The signal at 24.5 ppm in both these cases can be assigned to a phosphine coordinated precatalyst such as [RuCl2(NHC)(PPh3)(p-cymene)], similar to species Ia in Scheme 4. The other signals may correspond to various phosphine-coordinated Ru species including that of ruthenium-hydrido intermediates.
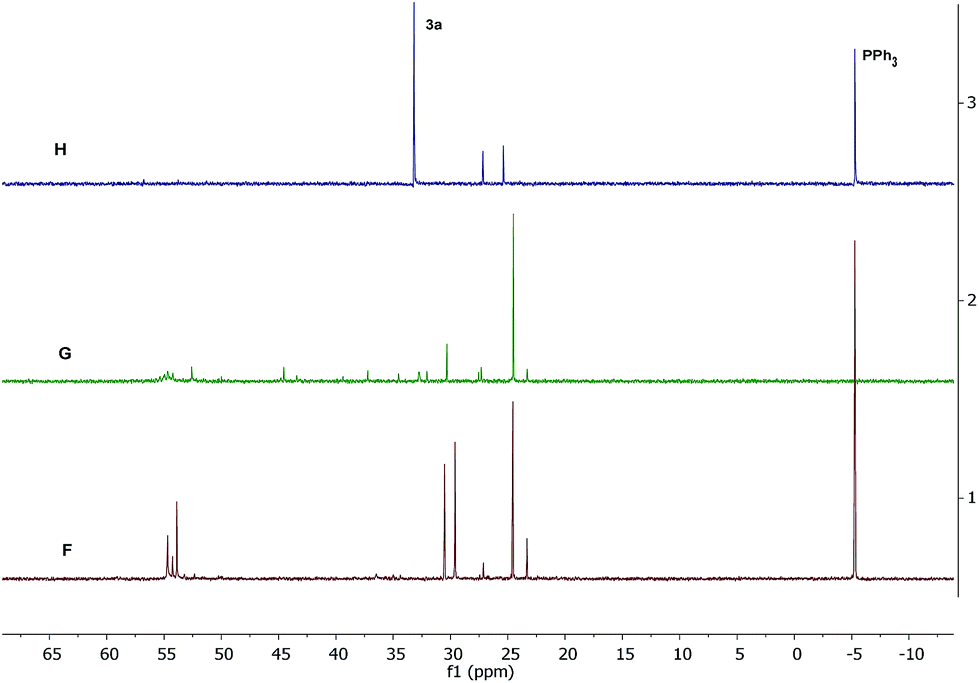 | ||
| Fig. 4 31P{1H} NMR spectroscopic analysis (162 MHz) for intermediate Ru species formed under reaction conditions with complex 1 + PPh3 (H, CDCl3), complex 3 + PPh3 (G, CDCl3) and complex 3a (F, DMSO-d6). | ||
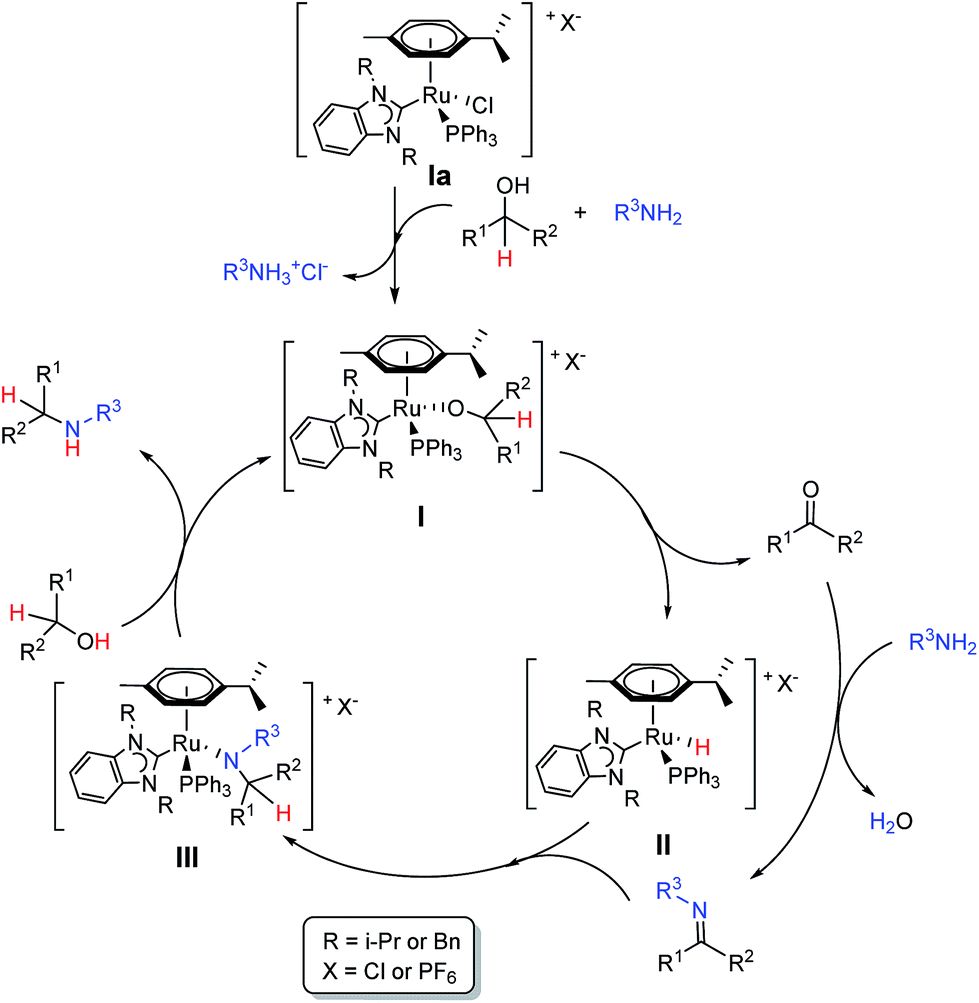 | ||
| Scheme 4 Proposed catalytic cycle. | ||
Based on the detection of various Ru–H species and Ru–P species as well as other literature reports42 we are proposing a simple catalytic cycle as shown in Scheme 3 likely involving a Ru–alkoxide complex I formed from the phosphine coordinated Ru–NHC pre-catalyst upon reaction with the alcohol, as the active catalytic species initiating the catalytic cycle. The Ru-complex I is transformed to a possible Ru–H complex II by elimination of the carbonyl intermediate, which condenses with the amine to form the imine intermediate. Insertion of this imine intermediate into the Ru–H species II generates the Ru-complex III, which produces the amine product and the active complex I upon subsequent reaction with the alcohol. PPh3 is optionally coordinated and in its absence, a corresponding neutral Ru–Cl complex or cationic complex with a vacant coordination site can be envisaged. Notably, all proposed catalytic intermediates are chiral-at-metal. In combination with the planar chirality resulting from restricted rotation of the p-cymene ligand, diastereomeric mixtures can be anticipated, which also explains the multiple species noted in Fig. 3 and 4. A detailed analysis, both experimental and theoretical calculations, is required to understand the exact nature of the various ruthenium intermediates formed under the reaction conditions to validate this proposed mechanism and is currently in progress at our laboratory.
Conclusions
In conclusion, we have reported the synthesis of ruthenium complexes bearing symmetrically (1, 3 and 3a) and unsymmetrically (2) substituted benzimidazolin-2-ylidene ligands. These ruthenium–NHC complexes showed promising catalytic activity for the alkylation of various amines using alcohols under solvent-free conditions in the absence of any external base. The catalysts were shown to be effective for the synthesis of pharmaceutically important amines such as fendiline, alverine, fenpiprane and prozapine giving generally very good yields. Five- to seven-membered N-heterocyclic amines were synthesized expediently from the corresponding diols using the cationic Ru catalyst 3a having both phosphine and carbene ligands. In general, the initial catalytic activity was found to be in the order 3a > 1 > 2 ⋙ 3. The robustness of these benzimidazolin-2-ylidene ruthenium complexes, their easy accessibility and wide applicability make them attractive catalysts for the synthesis of various important amines through alkylation using alcohols following hydrogen borrowing strategy. A catalytic cycle initiated by a Ru–alkoxide species is proposed based on the detection of Ru–H species by 1H and 31P NMR spectroscopic analysis.Acknowledgements
This work was funded by “GSK-EDB Singapore Partnership for Green and Sustainable manufacturing and the Institute of Chemical and Engineering Sciences (ICES), Agency for Science, Technology and Research (A*STAR), Singapore and National University of Singapore (NUS, WBS R143-000-513-592).Notes and references
- (a) Amino Group Chemistry: From Synthesis to the Life Sciences, ed. A. Ricci, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) Amines: Synthesis, Properties, and Applications, ed. S. A. Lawrence, Cambridge University, Cambridge, 2006 Search PubMed.
- R. N. Salvatore, C. H. Yoon and K. W. Jung, Tetrahedron, 2001, 57, 7785 CrossRef CAS.
- F. Alonso, P. Riente and M. Yus, Acc. Chem. Res., 2011, 44, 379 CrossRef CAS PubMed.
- P. J. Dunn, K. K. Hii and M. J. Krische, Sustainable Catalysis: Challenges and Practices for the Pharmaceutical and Fine Chemical Industries, John Wiley & Sons, Inc, 1st edn, 2013, pp. 121–137 Search PubMed.
- For reviews, see: (a) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555 CrossRef CAS; (b) T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753 RSC; (c) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681 CrossRef CAS PubMed; (d) G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611 CrossRef CAS PubMed; (e) S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853 CrossRef; (f) A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635 CrossRef CAS PubMed; (g) C. Gunanathan and D. Milstein, Science, 2013, 341, 249 CrossRef CAS PubMed; (h) M. Pera-Titus and F. Shi, ChemSusChem, 2014, 7, 720 CrossRef CAS PubMed.
- R. H. Crabtree, Organometallics, 2011, 30, 17 CrossRef CAS.
- R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, Tetrahedron Lett., 1981, 22, 4107 CrossRef CAS.
- (a) Y. Watanabe, Y. Tsuji and Y. Ohsugi, Tetrahedron Lett., 1981, 22, 2667 CrossRef CAS; (b) Y. Watanabe, Y. Tsuji, H. Ige, Y. Ohsugi and T. Ohta, J. Org. Chem., 1984, 49, 3359 CrossRef CAS; (c) Y. Tsuji, K.-T. Huh, Y. Ohsugi and Y. Watanabe, J. Org. Chem., 1985, 50, 1365 CrossRef CAS; (d) Y. Tsuji, K.-T. Huh and Y. Watanabe, Tetrahedron Lett., 1986, 27, 377 CrossRef CAS.
- (a) D. Gnanamgari, E. L. O. Sauer, N. D. Schley, C. Butler, C. D. Incarvito and R. H. Crabtree, Organometallics, 2009, 28, 321 CrossRef CAS; (b) D. Gnanamgari, C. H. Leung, N. D. Schley, S. T. Hilton and R. H. Crabtree, Org. Biomol. Chem., 2008, 6, 4442 RSC; (c) D. Balcells, A. Nova, E. Clot, D. Ganamangari, R. H. Crabtree and O. Eisenstein, Organometallics, 2008, 27, 2529 CrossRef CAS.
- (a) M. H. S. A. Hamid, C. L. Allen, G. W. Lamb, A. C. Maxwell, H. C. Maytum, A. J. A. Watson and J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766 CrossRef CAS PubMed; (b) A. J. Blacker, M. M. Farah, M. I. Hall, S. P. Marsden, O. Saidi and J. M. J. Williams, Org. Lett., 2009, 11, 2039 CrossRef CAS PubMed; (c) O. Saidi, A. J. Blacker, M. M. Farah, S. P. Marsden and J. M. J. Williams, Chem. Commun., 2010, 46, 1541 RSC; (d) A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, J. Org. Chem., 2011, 76, 2328 CrossRef CAS PubMed; (e) M. H. S. A. Hamid and J. M. J. Williams, Chem. Commun., 2007, 727 Search PubMed.
- (a) C. Gunanathan and D. Milstein, Angew. Chem., Int. Ed., 2008, 47, 8661 CrossRef CAS PubMed; (b) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588 CrossRef CAS PubMed.
- (a) S. Imm, S. Bähn, L. Neubert, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8126 CrossRef CAS PubMed; (b) S. Bahn, S. Imm, K. Mevius, L. Neubert, A. Tillack, J. M. J. Williams and M. Beller, Chem.–Eur. J., 2010, 16, 3590 CrossRef PubMed; (c) S. Imm, S. Bähn, M. Zhang, M. Neubert, H. Neumann, F. Klasovsky, J. Pfeffer, T. Haas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 7599 CrossRef CAS PubMed; (d) A. Tillack, D. Hollmann, D. Michalik and M. Beller, Tetrahedron Lett., 2006, 47, 8881 CrossRef CAS PubMed.
- (a) K.-I. Fujita, Y. Enoki and R. Yamaguchi, Tetrahedron, 2008, 64, 1943 CrossRef CAS PubMed; (b) R. Kawahara, K.-I. Fujita and R. Yamaguchi, J. Am. Chem. Soc., 2010, 132, 15108 CrossRef CAS PubMed; (c) R. Kawahara, K.-I. Fujita and R. Yamaguchi, Adv. Synth. Catal., 2011, 353, 1161 CrossRef CAS.
- Contributions on ruthenium catalysts from various groups: (a) S. P. Shan, T. T. Dang, A. M. Seayad and B. Ramalingam, ChemCatChem, 2014, 6, 808 CrossRef CAS; (b) D. Weickmann, W. Frey and B. Plietker, Chem.–Eur. J., 2013, 19, 2741 CrossRef CAS PubMed; (c) X. Cui, Y. Deng and F. Shi, ACS Catal., 2013, 3, 808 CrossRef CAS; (d) S. Agrawal, M. Lenormand and B. Martin-Matute, Org. Lett., 2012, 14, 1456 CrossRef CAS PubMed; (e) F. E. Fernandez, C. Puerta and P. Valerga, Organometallics, 2012, 31, 6868 CrossRef CAS; (f) R. Kawahara, K. Fujita and R. Yamaguchi, Adv. Synth. Catal., 2011, 353, 1161 CrossRef CAS; (g) R. Cano, D. J. Ramon and M. Yus, J. Org. Chem., 2011, 76, 5547 CrossRef CAS PubMed; (h) R. Kawahara, K.-I. Fujita and R. Yamaguchi, J. Am. Chem. Soc., 2010, 132, 15108 CrossRef CAS PubMed; (i) D. Pingen, C. Müller and D. Vogt, Angew. Chem., Int. Ed., 2010, 49, 8130 CrossRef CAS PubMed; (j) A. Arcelli, B. T. Khai and G. Porci, J. Organomet. Chem., 1982, 235, 93 CrossRef CAS; (k) A. Peeters, L. Claes, I. Geukens, I. Stassen and D. de Vos, Appl. Catal., A, 2014, 469, 191 CrossRef CAS PubMed; (l) M. Zhang, F. Xie, X. T. Wang, F. Yan, T. Wang, M. Chen and Y. Ding, RSC Adv., 2013, 3, 6022 RSC.
- Contributions on iridium catalysts from various groups: (a) D. Wang, X.-Q. Guo, C.-X. Wang, Y.-N. Wang, R. Zhong, X.-H. Zhu, L.-H. Cai, Z.-W. Gao and X.-F. Hou, Adv. Synth. Catal., 2013, 355, 1117 CrossRef CAS; (b) A. Wetzel, S. Wöckel, M. Schelwies, M. K. Brinks, F. Rominger, P. Hofmann and M. Limbach, Org. Lett., 2013, 15, 266 CrossRef CAS PubMed; (c) A. Bartoszewicz, R. Marcos, S. Sahoo, A. K. Inge, X. Zou and B. Martin-Matute, Chem.–Eur. J., 2012, 18, 14510 CrossRef CAS PubMed; (d) F. Li, H. Shan, L. Chen, Q. Kang and P. Zou, Chem. Commun., 2012, 48, 603 RSC; (e) H. Ohta, Y. Yuyama, Y. Uozumi and Y. M. A. Yamada, Org. Lett., 2011, 13, 3892 CrossRef CAS PubMed; (f) W. Zhang, X. Dong and W. Zhao, Org. Lett., 2011, 13, 5386 CrossRef CAS PubMed; (g) I. Cumpstey, S. Agrawal, K. E. Borbas and B. Martin-Matute, Chem. Commun., 2011, 47, 7827 RSC; (h) M. A. Berliner, S. P. A. Dubant, T. Makowski, K. Ng, B. Sitter, C. Wager and Y. Zhang, Org. Process Res. Dev., 2011, 15, 1052 CrossRef CAS; (i) S. Michlik and R. Kempe, Chem.–Eur. J., 2010, 16, 13193 CrossRef CAS PubMed; (j) B. Blanck, S. Michlik and R. Kempe, Adv. Synth. Catal., 2009, 351, 2903 CrossRef; (k) A. Prades, R. Corberan, M. Poyatos and E. Peris, Chem.–Eur. J., 2008, 14, 11474 CrossRef CAS PubMed; (l) Y.-H. Chang, Y. Nakajima and F. Ozawa, Organometallics, 2013, 32, 2210 CrossRef CAS; (m) P. Qu, C. Sun, J. Ma and F. Li, Adv. Synth. Catal., 2014, 356, 447 CrossRef CAS; (n) D. Wang, K. Zhao, X. Yu, H. Miao and Y. Ding, RSC Adv., 2014, 4, 42924 RSC; (o) J.-Q. Li and P. G. Anderson, Chem. Commun., 2013, 49, 6131 RSC.
- (a) N. Zotova, F. J. Roberts, G. H. Kelsall, A. S. Jessiman, K. Hellgardt and K. K. M. Hii, Green Chem., 2012, 14, 226 RSC; (b) T. Ishida, R. Takamura, T. Takei, T. Akita and M. Haruta, Appl. Catal., A, 2012, 261, 413 Search PubMed; (c) L. He, Y. Qian, R.-S. Ding, Y.-M. Liu, H.-Y. He and K.-N. Fan, ChemSusChem, 2012, 5, 621 CrossRef CAS PubMed; (d) C.-H. Tang, L. He, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Chem.–Eur. J., 2011, 17, 7172 CrossRef CAS PubMed; (e) L. He, X. B. Lou, J. Ni, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Chem.–Eur. J., 2010, 16, 13965 CrossRef CAS PubMed.
- (a) H. Liu, G. K. Chuah and S. Jaenicke, J. Catal., 2012, 292, 130 CrossRef CAS PubMed; (b) K. Shimizu, K. Shimura, M. Nishimura and A. Satsuma, RSC Adv., 2011, 1, 1310 RSC; (c) X. Cui, Y. Zhang, F. Shi and Y. Deng, Chem.–Eur. J., 2011, 17, 1021 CrossRef CAS PubMed; (d) K. Shimizu, M. Nishimura and A. Satsuma, ChemCatChem, 2009, 1, 497 CrossRef CAS; (e) I. Geukens, F. Vermoortele, M. Meledina, S. Turner, G. van Tendeloo and D. E. de Vosa, Appl. Catal., A, 2014, 469, 373 CrossRef CAS PubMed.
- M. Bertoli, A. Choualeb, A. J. Lough, B. Moore, D. Spasyuk and D. G. Gusev, Organometallics, 2011, 30, 3479 CrossRef CAS.
- P. Satyanarayana, G. M. Reddy, H. Maheswaran and M. L. Kantam, Adv. Synth. Catal., 2013, 355, 1859 CrossRef CAS.
- (a) T. T. Dang, B. Ramalingam, S. P. Shan and A. M. Seayad, ACS Catal., 2013, 3, 2536 CrossRef CAS; (b) M. Ousmane, G. Perrussel, Z. Yan, J.-M. Clacens, F. de Campo and M. Pera-Titus, J. Catal., 2014, 309, 439 CrossRef CAS PubMed; (c) F. Ozawa, H. Okamoto, S. Kawagishi, S. Yamamoto, T. Minami and M. Yoshifuji, J. Am. Chem. Soc., 2002, 124, 10968 CrossRef CAS PubMed; (d) S.-C. Yang and C.-W. Hung, J. Org. Chem., 1999, 64, 5000 CrossRef CAS; (e) A. Corma, T. Ródenas and M. Sabater, Chem.–Eur. J., 2010, 16, 254 CrossRef CAS PubMed; (f) Y. Q. X. Zhang, X. Cui, F. Shi and Y. Deng, Tetrahedron Lett., 2011, 52, 1334 CrossRef CAS PubMed; (g) Y. Shiraishi, K. Fujiwara, Y. Sugano, S. Ichikawa and T. Hirai, ACS Catal., 2013, 3, 312 CrossRef CAS; (h) A. Martinez-Asencio, M. Yus and D. J. Ramón, Synthesis, 2011, 3730 CAS.
- (a) Y. S. Zhao, S. W. Foo and S. Saito, Angew. Chem., Int. Ed., 2011, 50, 3006 CrossRef CAS PubMed; (b) R. Martinez, D. J. Ramón and M. Yus, Org. Biomol. Chem., 2009, 7, 2176 RSC.
- (a) M. Dixit, M. Mishra, P. A. Joshi and D. O. Shah, Catal. Commun., 2013, 33, 80 CrossRef CAS PubMed; (b) A. Martinez-Asencio, D. J. Ramón and M. Yus, Tetrahedron, 2011, 67, 3140 CrossRef CAS PubMed; (c) P. R. Likhar, R. Arundhathi, M. L. Kantam and P. S. Prathima, Eur. J. Org. Chem., 2009, 5383 CrossRef CAS; (d) F. Shi, M. K. Tse, X. Cui, D. Gördes, D. Michalik, K. Thurow, Y. Deng and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 5912 CrossRef CAS PubMed; (e) F. Santoro, R. Psaro, N. Ravasio and F. Zaccheria, RSC Adv., 2014, 4, 2596 RSC.
- (a) K. Shimizu, N. Imaiida, K. Kon, S. M. A. H. Siddiki and A. Satsuma, ACS Catal., 2013, 3, 998 CrossRef CAS; (b) K. Shimizu, K. Kon, W. Onodera, H. Yamazaki and J. N. Kondo, ACS Catal., 2013, 3, 112 CrossRef CAS.
- H. Quin, N. Yamagiwa, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2007, 46, 409 CrossRef PubMed.
- J.-M. Yang, R. Jiang, L. Wu, X.-P. Xu, S.-Y. Wang and S.-J. Ji, Tetrahedron, 2013, 69, 7988 CrossRef CAS PubMed.
- A. Abdukader, H. Jin, Y. Cheng and C. Zhu, Tetrahedron Lett., 2014, 55, 4172 CrossRef CAS PubMed.
- (a) A. J. Arduengo, H. V. R. Dias, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1992, 114, 5530 CrossRef CAS; (b) W. A. Herrmann and C. Kocher, Angew. Chem., Int. Ed., 1997, 36, 2162 CrossRef CAS; (c) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; (d) M. F. Lappert, J. Organomet. Chem., 1988, 358, 185 CrossRef CAS; (e) D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef CAS PubMed; (f) M. S. Sanford, J. A. Love and R. H. Grubbs, Organometallics, 2001, 20, 5314 CrossRef CAS; (g) N. M. Scott and S. P. Nolan, Eur. J. Inorg. Chem., 2005, 1815 CrossRef CAS; (h) F. Glorius, N-Heterocyclic Carbenes, Transition Metal Catalysis, Springer, Berlin, 2007, vol. 21 Search PubMed.
- A. Pontes da Costa, M. Viciano, M. Sanaú, S. Merino, J. Tejeda, E. Peris and B. Royo, Organometallics, 2008, 27, 1305 Search PubMed.
- A. Prades, M. Viciano, M. Sanaú and E. Peris, Organometallics, 2008, 27, 4254 Search PubMed.
- (a) F. E. Fernández, E. M. C. Puerta and P. Valerga, Organometallics, 2012, 31, 6868 Search PubMed; (b) F. E. Fernández, M. C. Puerta and P. Valerga, Organometallics, 2011, 30, 5793 Search PubMed.
- L. Oehninger, M. Stefanopoulou, H. Alborzinia, J. L. Schur, S. Namikawa, A. Muñoz-Castro, R. W. Köster, K. Baumann, S. Wölfl, W. S. Sheldrick and I. Ott, Dalton Trans., 2013, 42, 1657 Search PubMed.
- (a) H. V. Huynh, L. R. Wong and S. P. Ng, Organometallics, 2008, 27, 2231 Search PubMed; (b) H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395 Search PubMed.
- J. H. Dam, G. Osztrovoszky, L. U. NordstrØm and R. Madsen, Chem.–Eur. J., 2010, 16, 6820 Search PubMed.
- N. Gürbüz, S. Yaşar, E. Ö. Özcan, İ. Özdemir and B. Çetinkaya, Eur. J. Inorg. Chem., 2010, 3051 Search PubMed.
- Similar results were observed with P(Cy)3 and PPh3 as the additive. Hence PPh3 was selected for further evaluation.
- C. R. Jan, K. C. Lee, K. J. Chou, J. S. Cheng, J. L. Wang, J. L. Wang, Y. K. Lo, H. T. Chang, K. Y. Tang, C. C. Yu and J. K. Huang, Cancer Chemother. Pharmacol., 2001, 48, 37 Search PubMed.
- [Cp*IrI2]2 catalyzed direct synthesis of fendiline was reported by the alkylation of 1-phenylethylamine using 3,3-diphenyl-1-propanol as alkylating agent with 85% isolated yield in aqueous medium. See ref. 10c.
- T. Wittmann, L. Paradowski, P. Ducrotte, L. Bueno and M.-C. A. Delestrain, Aliment. Pharmacol. Ther., 2010, 31, 615 Search PubMed.
- S. Li, K. Huang, J. Zhang, W. Wu and X. Zhang, Org. Lett., 2013, 15, 1036 Search PubMed.
- K.-I. Fujita, T. Fuji and R. Yamaguchi, Org. Lett., 2004, 6, 3525 Search PubMed.
- Y. Tsuji, K.-T. Huh, Y. Ohsugi and Y. Watanabe, J. Org. Chem., 1985, 50, 1365 Search PubMed.
- (a) X. Ye, P. N. Plessow, M. K. Brinks, M. Schelwies, T. Schaub, F. Rominger, R. Paciello, M. Limbach and P. Hofmann, J. Am. Chem. Soc., 2014, 136, 5923 Search PubMed; (b) D. Pingen, M. Lutz and D. Vogt, Organometallics, 2014, 33, 1623 Search PubMed; (c) C. Y. Song, S. L. Qu, Y. Tao, Y. F. Dang and Z. X. Wang, ACS Catal., 2014, 4, 2854 Search PubMed; (d) H. X. Li, X. T. Wang, M. W. Wen and Z. X. Wang, Eur. J. Inorg. Chem., 2012, 5011 Search PubMed.
- NMR identification of some of the similar Ru–H species: (a) K. Umezawa-Vizzini and T. R. Lee, Organometallics, 2004, 23, 1448 Search PubMed; (b) P. Buskens, D. Giunta and W. Leitner, Inorg. Chim. Acta, 2004, 357, 1969 Search PubMed; (c) P. A. van der Schaaf, R. Kolly and A. Hafner, Chem. Commun., 2000, 1045 Search PubMed; (d) C. P. Laua, S. M. Nga, G. Jia and Z. Lin, Coord. Chem. Rev., 2007, 251, 2223 Search PubMed; (e) V. L. Chantler, S. L. Chatwin, R. F. R. Jazzar, M. F. Mahon, O. Saker and M. K. Whittlesey, Dalton Trans., 2008, 2603 Search PubMed; (f) S. Burling, B. M. Paine, D. Nama, V. S. Brown, M. F. Mahon, T. J. Prior, P. S. Pregosin, M. K. Whittlesey and J. M. J. Williams, J. Am. Chem. Soc., 2007, 129, 1987 Search PubMed; (g) M. Arisawa, Y. Terada, K. Takahashi, M. Nakagawa and A. Nishida, J. Org. Chem., 2006, 71, 4255 Search PubMed; (h) S. Burling, G. Kociok-Kohn, M. F. Mahon, M. K. Whittlesey and J. M. J. Williams, Organometallics, 2005, 24, 5868 Search PubMed; (i) C. L. Lund, M. J. Sgro, R. Cariou and D. W. Stephan, Organometallics, 2012, 31, 802 Search PubMed.
- See ESI† for details.
- For NMR characterization of some of the representative H–Ru–P complexes, see: (a) C. S. Yi, T. N. Zeczycki and I. A. Guzei, Organometallics, 2006, 25, 1047 Search PubMed; (b) F. Kakiuchi, T. Kochi, E. Mizushima and S. Murai, J. Am. Chem. Soc., 2010, 132, 17741 Search PubMed; (c) T. Koreeda, T. Kochi and F. Kakiuchi, Organometallics, 2013, 32, 682 Search PubMed; (d) A. E. W. Ledger, P. A. Slatford, J. P. Lowe, M. F. Mahon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 716 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1028107. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra15398g |
| ‡ Indian Institute of Science Education and Research, Dr Homi Bhabha Road, Pashan, Pune 411008, India. |
| This journal is © The Royal Society of Chemistry 2015 |