
Enhanced electrochemical performance of LiMn2O4 cathode with a Li0.34La0.51TiO3-coated layer†
Abstract
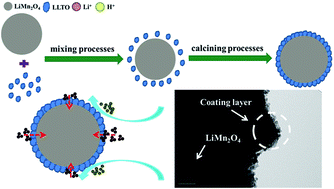
Spinel cathode materials consisting of LiMn2O4@Li0.34La0.51TiO3 (LMO@LLTO) have been synthesized by a new and facile solid-phase route in air. When used as cathode for lithium ion batteries, the LLT01 (LiMn2O4 with 20 nm Li0.34La0.51TiO3 coating) sample tested at 1 C rate exhibits 9.2% capacity loss and 90.4% capacity retention after 200 cycles at 25 °C and 55 °C. The improved cycling performance of composites is attributed to the LLT01 coating on the surface of spinel particles. The polycrystalline LLTO-coated layer could provide superior ionic conductivity and prevent Mn dissolution in electrolyte during electrochemical cycling.
 Please wait while we load your content...
Please wait while we load your content...

