
Preparation of porous carbon microspheres anode materials from fine needle coke powders for lithium-ion batteries
Wenfeng Renab,
Zailei Zhanga,
Yanhong Wang*a,
Guangwei Kana,
Qiangqiang Tan*a,
Ziyi Zhongc and
Fabing Su*a
aState Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, China 100190. E-mail: wangyanhong@ipe.ac.cn; qtan@ipe.ac.cn; fbsu@ipe.ac.cn; Fax: +86-10-82544851; Tel: +86-10-82544850
bUniversity of Chinese Academy of Sciences, Beijing, China 100049
cInstitute of Chemical Engineering and Sciences, A*star, 1 Pesek Road, Jurong Island, Singapore 627833
First published on 8th January 2015
Abstract
A large amount of fine carbon powders (graphite and cokes) are generated as the solid waste in the manufacture of carbon-based materials in industry. How to utilize these abundant powders to generate products with high value still remains a big challenge. Herein, we report the preparation of porous carbon microspheres (PCMs) employing waste non-graphitized needle coke and graphitized needle coke as the fine carbon powder representatives, demonstrating their use as anode materials for lithium-ion batteries. It was found that the graphitized PCMs had a size of 8–30 μm and surface areas between 50 and 120 m2 g−1. When used as the anode materials, their charge capacity at the current density of 50 mA g−1 was comparable to that of the commercial graphite microspheres, but they exhibited higher rate performance.
1. Introduction
Needle coke (NC) generated from coal, petroleum or pitch coke (PC), and synthetic graphite (SG) from NC or PC as well as natural graphite (NG) are widely employed as raw carbon-based materials to produce various carbon electrodes for batteries and electrolysis devices.1,2 In the manufacturing processes of these carbon materials such as pulverization, purification, modification, molding, shaping, and graphitization, there is always the generation of large amounts of carbon-based fine powders as solid wastes, which are usually combusted as cheap solid fuel or directly abandoned. How to effectively utilize these fine carbon waste powders to generate high economic value while minimizing environmental pollution is of great interest but still remains as a big challenge.Potentially, these waste carbon materials can be used to prepare lithium-ion batteries anodes, as the commercial anodes in lithium-ion batteries are also predominantly made from carbon materials such as abundant NG, SG, PC, and NC.3–5 However, making carbon anodes from these fine carbon powders needs to overcome more technical barriers, e.g., the graphitic degree should be increased, and the powder should be casted into certain shape with a proper size. In addition, even for the commercial carbon anodes, their electrochemical performances such as capacity, coulombic efficiency, cycling stability, and rate performance are proven to depend on their preparation conditions and microstructures, and there is still ample room for further improvement. For example, Chen et al.6 and Ma et al.7 found that the pretreatment was important in the formation of solid electrolyte interphase (SEI) film during cycling process for carbon anodes made of PC and NC particles. It was found that the electrochemical performances of NG could be improved by the mild surface oxidation in air8 or by surface coating of carbon via thermal decomposition of C2H2.9 Or Yoshio et al. also observed that the initial coulombic efficiency of spherical NG particles was increased by coated carbon derived from the thermal vapor decomposition of toluene,10,11 because the coated carbon could keep the NG surface from direct contact with the electrolyte, resulting in less exfoliation and decomposition of the electrolyte. Some other examples of surface carbon coating include coating SG with amorphous carbon (generated from glucose),12 conductive polypyrrole13 or poly(vinyl chloride) powders.14 Also, the incorporation of Si particles could enhance the capacity of NG microspheres.15,16
In addition, recent work demonstrated that the electrochemical performance of carbon anodes could be enhanced by forming conductive carbon networks within the carbon anodes via incorporating nanostructured hollow carbon,17,18 carbon nanotubes,19 carbon nanofibers,20 graphene,21 and hard carbon (HC) network,22 and also by introduction of porous structures.23–26 Comparing with the conventional carbon materials, these inner carbon nanostructures provide shorter diffusion length for Li ion transport, more active sites for Li-ion storage, as well as reduce stress induced by volume changes. However, even for the commercial graphite microspheres (GMs) used as anodes, which are normally manufactured by spheroidizing SG or NG microparticles followed with surface carbon coating treatment, there is no presence of any porous structure inside, thus their rate performance is usually low. However, their spherical morphology is most desirable for high packing density and good mobility during electrode fabrication.
The spray drying technique has been widely used to granulate many pharmaceutical and chemical products,27,28 such as polymer microspheres,29 hollow spherical silica nanoparticles,30 mannitol particles,31 spherical hollow metal–organic frameworks,32 porous carbon microspheres,22 and spherical Si/C composite spheres.33 The industrial process using this technique is well established and can be adaptable to the production of microspherical materials that require sophisticated nanostructures as well as large quantity syntheses.
In this work, in order to develop the carbon anodes from the waste fine carbon powders, we employed the spray-drying method to make the porous carbon microspheres (PCMs) from the representative fine NC and graphitized NC (GNC) powders using sucrose as the binder. The prepared PCMs have not only the preferred spherical morphology, but also the well-developed inner porous structure and HC network, very obvious structural characteristics conducive to the high performance carbon anodes. The whole preparation process is illustrated in Fig. 1. In the first step, the fine NC powders (Fig. 1a) were dispersed in sucrose solution (as binder) and spray-dried to collect PCMs (Fig. 1b), which were subsequently carbonized at 900 °C and graphitized at 2800 °C to obtain PCM-900 and PCM-2800 samples (Fig. 1c and d), respectively. In the second step, the fine NC powders were firstly graphitized to create GNC (Fig. 1e), and then similarly followed with spray drying process to achieve graphitized PCMs (GPCMs) (Fig. 1f) and carbonization process at 900 °C to obtain GPCM-900 sample (Fig. 1g). It is found that the capacity of graphitized microspheres (PCM-2800 and GPCM-900) is comparable to that of GMs but with much better rate performance. The work demonstrates the high technical feasibility of making microspherical carbon-based anode materials in lithium-ion batteries from graphitized and non-graphitized fine carbon powders.
 | ||
| Fig. 1 Illustration of the preparation process of PCMs. | ||
2. Experimental
2.1 Material synthesis
Commercial NC powders (Shanghai Hongte chemical Co., Ltd., China) were sieved to obtain fine NC powders (≤10 μm), which were further graphitized under vacuum at 2800 °C for 12 hours to obtain GNC powders.22 As illustrated in Fig. 1, a given amount of NC/GNC and sucrose (A.R., Sinopharm Chemical Reagent Co., Ltd.) (NC/GNC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :sucrose:H2O = 1:1:10, wt%) was mixed under ultrasonication for 30 min, which was subsequently sprayed via spray drying machine (YC-015, Shanghai Pilotech Instrument and Equipment Co., Ltd., China). Then the as-prepared precursors (denoted as PCMs and GPCMs) were collected. Finally, the precursors were calcined in nitrogen at 900 °C for 2 hours (denoted as PCM-900 and GPCM-900). In addition, the as-prepared PCMs precursor was graphitized under vacuum at 2800 °C for 12 hours to obtain PCM-2800.
:sucrose:H2O = 1:1:10, wt%) was mixed under ultrasonication for 30 min, which was subsequently sprayed via spray drying machine (YC-015, Shanghai Pilotech Instrument and Equipment Co., Ltd., China). Then the as-prepared precursors (denoted as PCMs and GPCMs) were collected. Finally, the precursors were calcined in nitrogen at 900 °C for 2 hours (denoted as PCM-900 and GPCM-900). In addition, the as-prepared PCMs precursor was graphitized under vacuum at 2800 °C for 12 hours to obtain PCM-2800.
2.2 Characterization
X-ray diffraction patterns (XRD) were recorded on a PANalytical X'Pert PRO MPD using the Kα radiation of Cu (λ = 1.5418 Å). The microscopic features of the sample were observed by field-emission scanning electron microscopy (SEM) (JSM-7001F, JEOL, Tokyo, Japan) and transmission electron microscopy (TEM) (JEM-2010F, JEOL, Tokyo, Japan). For SEM analysis of cycled electrodes, the cell was disassembled in the argon-filled glove box, and then the cycled electrodes were washed by dimethyl carbonate and were dried overnight at the room temperature under vacuum. The porous property of the samples was investigated using physical adsorption of nitrogen at liquid-nitrogen temperature (−196 °C) on an automatic volumetric sorption analyzer (NOVA3200e, Quantachrome). Prior to the measurement, the sample was degassed at 200 °C for 24 h under vacuum. The specific surface areas were determined according to the Brunauer–Emmett–Teller (BET) method in the relative pressure range of 0.05–0.2. The tap density of samples was measured with a standard cylinder marked with a volume value. The particle size distribution (PSD) was measured using a laser particle size analyzer (Model BT-9300Z, Better size Instruments, Ltd., China). A Raman spectroscopy (Renishaw inVia plus, England) with an excitation wavelength of 514.5 nm and a beam spot size of 1–2 μm was used to characterize sample. Thermogravimetric (TG) analysis was carried out on an EXSTAR TG/DTA 6300 (Seiko Instruments, Japan) using a heating rate of 10 °C min−1 in air (200 mL min−1).2.3 Electrochemical measurement
The working electrode was prepared by mixing the active materials, acetylene black, and polyvinylidene fluoride (PVDF) in a weight ratio of 80:10:10 with N-methylpyrrolidone (NMP) as a solvent. The resulting slurries were cast onto copper current collectors. The foils were rolled into 25 μm thin sheets, and then dried at 40 °C for 24 h. The foils were cut into disks which were 14 mm in diameter, and then dried at 120 °C under vacuum for 24 h. CR2016 coin-type cells were assembled in an argon-filled glove box with lithium foils as the counter electrodes and polypropylene macroporous films (Celgard 2400) as separators. The liquid electrolyte was 1 mol L−1 LiPF6 in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (1:1, v/v). The galvanostatic charge and discharge tests were carried out by the NEWARE-BTS-5 V/10 mA testing instrument (Neware Co., Ltd., Shenzhen, China) in a voltage range between 0.005 and 2.0 V at the current rates of 50, 100, 500, and 1000 mA g−1. Cyclic voltammetry tests were carried out between 0.005 and 2.0 V at a scan rate of 0.1 mV s−1 using CHI660D electrochemical workstation. Electrochemical impedance spectroscopy (EIS) measurements were carried out on CHI660D electrochemical workstation in the frequency range from 100 kHz to 10 mHz at an ac-oscillation of 5 mV. All electrochemical performance were measured at room temperature. The commercial GMs (Qingdao Tianhe Graphite Co., Ltd., China) were employed for the electrochemical comparison.
3. Results and discussion
Fig. 2 shows the SEM images of NC, GNC, PCM-900, PCM-2800, and GMs. It is seen that NC (Fig. 2a) and GNC (Fig. 2b) have a flaky morphology with a size of several micrometers and a thickness of several hundred nanometers. PCM-900 (Fig. 2c), PCM-2800 (Fig. 2d) and GPCM-900 (not shown here) have microspherical morphology with a diameter of 8–30 μm, suggesting that the spherical morphology of PCMs can be well maintained after the heat treatment at 900 and 2800 °C. The SEM image of single PCM-2800 microsphere in Fig. 2e clearly shows that the microspheres are assembled with fine GNC powders. GMs (Fig. 2f) shows ellipsoidal morphology with a size of 10–20 μm. Table 1 compiles the physical properties of all the samples. It can be seen that the measured surface areas of NC, GNC, PCM-900, PCM-2800, GPCM-900, and GMs are 25.6, 19.4, 122.3, 50.2, 90.5, and 10.1 m2 g−1, respectively. The surface areas of PCM-900 and GPCM-900 are higher than NC and GNC, respectively, mainly due to the introduction of porous structure derived from interspace among NC/GNC particles and micropores within HC generated from the sucrose pyrolysis. The surface areas of GNC and PCM-2800 are lower than that of NC and PCM-900 respectively because the graphitization process normally results in the shrinkage and higher density of carbon materials. In addition, the tap densities of PCM-900, PCM-2800, and GPCM-900 in Table 1 are in the range of 0.47–0.56 g mL−1, less than that of GMs (1.18 g mL−1) due to the presence of their inner porous structure in PCMs, but much larger than that of NC (0.25 g mL−1) and GNC (0.23 g mL−1) indicating the spray drying process makes NC or GNC packed tightly.34 However, for PCMs, their special surface areas are a little larger and their tap densities seem to be lower than those of commercial graphite anodes, which may result in the worse cycling performance and low volume capacity, respectively. These problems could be solved by filling more active materials with a higher capacity (Si or transition metal oxide nanoparticles) and by coating carbon via chemical vapor deposition into the inner pores of PCMs. | ||
| Fig. 2 SEM images of NC (a), GNC (b), PCM-900 (c), PCM-2800 (d and e), and GMs (f). | ||
| Sample | SBETa | TDb | PSc | 2θ(002)d | d002e | Lcf | ID/IGg |
|---|---|---|---|---|---|---|---|
| a SBET, (m2 g−1), BET surface area measured by N2 adsorption isotherm.b TD, (g mL−1), tap density.c PS, (μm), particle size derived from the maximum peak position of the particle size distribution curves.d 2θ(002), (degree), the position of (002) peak from the XRD pattern.e d002, (nm), the interlayer spacing distance between the adjacent graphene layers calculated from the (002) peak of XRD pattern using Bragg's formula d002 = λ/2sinθ, where, λ is the wavelength of Cu-Ka radiation, and θ is the angle of (002) peak, λ = 1.5418 Å.f Lc, (nm), crystallite sizes along the c axis, calculated from the (002) peak of XRD pattern using Scherrer's formula (Lc = Kλ/βcosθ), where, K = 0.9, and β is the width of (002) peak at half height.g ID/IG, the ratio of the relative intensities of D- and G-band peaks. |
|||||||
| NC | 25.6 | 0.23 | 2.0 | 25.5 | 0.349 | 4.9 | 1.26 |
| GNC | 19.4 | 0.25 | 3.0 | 26.6 | 0.335 | 20.7 | 0.21 |
| PCM-900 | 122.3 | 0.47 | 14.6 | 25.8 | 0.345 | 4.6 | 1.25 |
| PCM-2800 | 50.2 | 0.56 | 15.8 | 26.5 | 0.336 | 19.6 | 0.36 |
| GPCM-900 | 90.5 | 0.56 | 15.7 | 26.6 | 0.335 | 16.3 | 0.56 |
| GMs | 10.1 | 1.18 | 22.2 | 26.5 | 0.336 | 36.5 | 0.17 |
Fig. 3 shows the TEM images of NC, GNC, PCM-2800, and GMs. It is seen that NC (Fig. 3a) is amorphous but GNC (Fig. 3b) is well graphitized. The regular lattice plane distance between the straight layers within GNC is measured to be around 0.335 nm, close to that of graphite. The TEM image of PCM-2800 in Fig. 3c shows the presence of amorphous HC, which is coated on the surface of flaky GNC. This is because HC is non-graphitized but NC is graphitized. For GMs (Fig. 3d), the typical (002) planes with a distance of 0.336 nm are observed. Thus, it is concluded that both NC and PCM-900 have a lower graphitization degree, while PCM-2800 and GPCM-900 are composed of flaky GNC, having a higher graphitization degree, comparable to that of GMs, which are further confirmed by below XRD and Raman results.
 | ||
| Fig. 3 TEM images of NC (a), GNC (b), PCM-2800 (c), and GMs (d). | ||
Fig. 4a shows the PSD curves of all the samples. It can be seen that the particle sizes of NC and GNC are in the range of 1–10 μm, consistent with the above SEM observation. After spray drying and carbonization or graphitization, the particle sizes of PCM-900, PCM-2800, and GPCM-900 are in the range of 5–30, 5–30, and 5–35 μm, respectively, comparable to that of GMs (10–35 μm). The maximum peak positions of these PSD curves are at 14.6, 15.8, 15.7, and 22.2 μm, respectively, which are in a comparable size range. However, the size ranges of PCM-900, PCM-2800, and GPCM-900 are much wider than that of GMs (also see above SEM images). This is because of the limitation of the spray drying machine used with the lab scale. It is believed if the spray drying process is conducted using industrial-scale machine, the size range of obtained microspheres will be narrowed.22
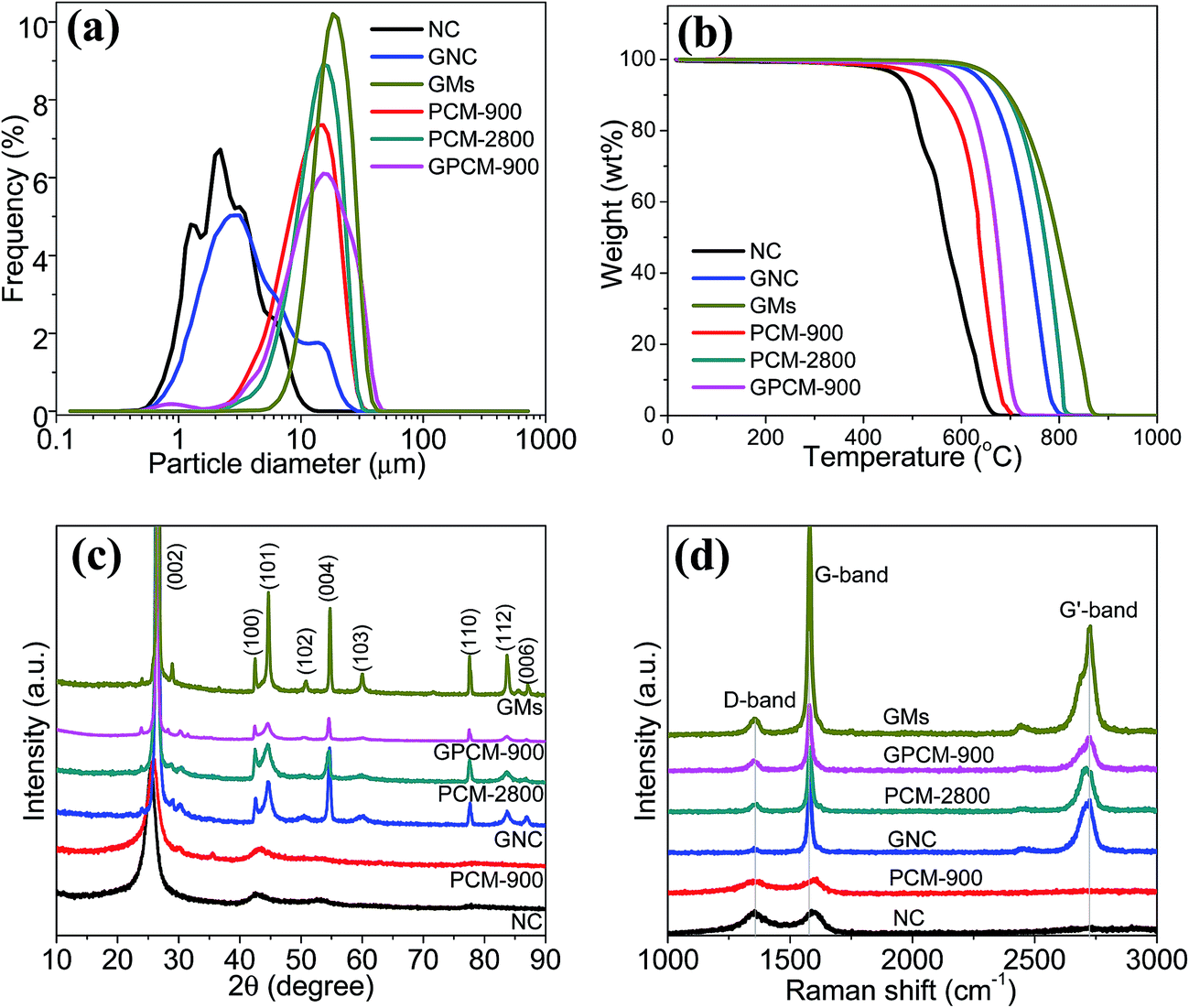 | ||
| Fig. 4 The PSD curves (a), TG curves (b), XRD patterns (c), and Raman spectra (d) of all the samples. | ||
Fig. 4b shows the TG curves of all the samples measured in air. The weight loss of NC derived from carbon combustion is located in the range of 500–670 °C, lower than that of PCM-900 (600–700 °C) due to the fact that NC particles are more densely packed with HC, leading to the less active sites exposed to oxygen. The lower combustion temperature of GPCM-900 (650–720 °C) than that of GNC (700–800 °C) stems from the introduction of amorphous HC that normally has a lower combustion temperature than graphitized carbon.35 The higher combustion temperature of GNC and PCM-2800 than that of NC and PCM-900 respectively is mainly because of the higher graphitization degree of the former two.1 As reported, the combustion/oxidation of carbon materials preferentially takes place at the defects in graphene layer, the edge of the pores, and the disordered graphene layers.35 Among all the samples, GMs have the highest combustion temperature range (750–880 °C).
Fig. 4c shows the XRD patterns of all the samples. NC and PCM-900 have two weak diffraction peaks at 2θ values of ∼25.5 and ∼42.9°. For GNC, PCM-2800, GPCM-900, and GMs, the diffraction peaks at 2θ values of 26.5, 42.5, 44.7, 50.8, 54.8, 60.0, 77.6, and 83.7° are assigned to (002), (100), (101), (102), (004), (103), (110), and (112) planes of graphitic carbon (JCPDS no. 865-6212), respectively.10 The (002) peak shifts to the higher degree from NC and PCM-900 to GNC, PCM-2800, and GPCM-900, indicating the decrease of layer spaces of graphite and the increase of their graphitic crystallinity, consistent with the above TEM analysis. The crystallite size along c-axis (Lc) is calculated to be around 4.9 nm for NC, and 4.6 nm for PCM-900, 20.7 nm for GNC, 19.6 nm for PCM-2800, 16.3 nm for GPCM-900, and 36.5 nm for GMs as shown in Table 1. These results suggest NC and PCM-900 have much lower crystallinity degree than GNC, PCM-2800, GPCM-900, and GMs. Fig. 4d displays the Raman spectra of all the samples. It is observed that there are two peaks for NC and PCM-900 but three peaks for GNC, PCM-2800, GPCM-900, and GMs. The weak peak at about 1350 cm−1 (D-band) is associated with the vibration of carbon atoms with dangling bonds in plane terminations of the disordered graphite that is related to amorphous carbon materials, while the peak at 1580 cm−1 (G band) is related to the vibration of sp2-bonded carbon atoms in a two-dimensional hexagonal lattice, resulting from the stretching modes of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of typical graphite.36,37 The low-intensity and weak G-band peak suggests a structural imperfection of the graphene sheets such as defects and the small crystal domain size, which can be further characterized with the ratio of the relative intensities of D- and G-band peaks (ID/IG). The small ID/IG value indicates that the carbon material is composed of ordered graphene sheets with a high graphitization nature.37 The values of ID/IG for NC, GNC, PCM-900, PCM-2800, GPCM-900, and GMs in Table 1 are 1.26, 0.21, 1.25, 0.36, 0.56, and 0.17, respectively, suggesting that except for GMs, GNC has the highest graphitization degree. The lower ID/IG values for PCM-2800 and GPCM-900 is due to the introduction of amorphous HC. These results are consistent with the above XRD analysis. In addition, for GNC, PCM-2800, GPCM-900, and GMs, there is a G′-band peak at around 2700 cm−1,38 which is another typical characteristic of undisturbed or highly ordered graphitic lattices, implying GNC, PCM-2800, and GPCM-900 have a higher graphitization degree than NC and PCM-900. All of these results suggest the graphitic crystallinity order is GMs > GNC > PCM-2800 > GPCM-900 > PCM-900 > NC.
C bonds of typical graphite.36,37 The low-intensity and weak G-band peak suggests a structural imperfection of the graphene sheets such as defects and the small crystal domain size, which can be further characterized with the ratio of the relative intensities of D- and G-band peaks (ID/IG). The small ID/IG value indicates that the carbon material is composed of ordered graphene sheets with a high graphitization nature.37 The values of ID/IG for NC, GNC, PCM-900, PCM-2800, GPCM-900, and GMs in Table 1 are 1.26, 0.21, 1.25, 0.36, 0.56, and 0.17, respectively, suggesting that except for GMs, GNC has the highest graphitization degree. The lower ID/IG values for PCM-2800 and GPCM-900 is due to the introduction of amorphous HC. These results are consistent with the above XRD analysis. In addition, for GNC, PCM-2800, GPCM-900, and GMs, there is a G′-band peak at around 2700 cm−1,38 which is another typical characteristic of undisturbed or highly ordered graphitic lattices, implying GNC, PCM-2800, and GPCM-900 have a higher graphitization degree than NC and PCM-900. All of these results suggest the graphitic crystallinity order is GMs > GNC > PCM-2800 > GPCM-900 > PCM-900 > NC.
Fig. 5a shows the initial discharge–charge curves of all the samples at the current density of 50 mA g−1 between 0.005 and 2.0 V. The initial discharge capacity is 477 mA h g−1 for NC, 475 mA h g−1 for PCM-900, 433 mA h g−1 for GNC, 394 mA h g−1 for PCM-2800, 416 mA h g−1 for GPCM-900, and 412 mA h g−1 for GMs, while their initial charge capacities are 241, 266, 332, 351, 338, and 354 mA h g−1, respectively, as shown in Table 2. Thus, the initial coulomb efficiency of the samples is in the order of PCM-2800 (89.2%) > GMs (85.8%) > GPCM-900 (81.4%) > GNC (76.7%) > PCM-900 (56.0%) > NC (50.5%). The much lower initial coulombic efficiency of non-graphitized carbon (NC and PCM-900) than those of the graphitized carbon (GNC, PCM-2800, GPCM-900, and GMs) may be because there are many defects on the surface and internal porosities in the non-graphitized carbon, which raise more secondary reactions involving electrolyte decomposition between electrode and electrolyte resulting in a high irreversibility (low columbic efficiency).4 As a result, more SEI films will be generated as more lithium ions will participate in an irreversible reaction, corresponding to the discharge plateaus at about 0.70 V for NC and PCM-900 in the initial cycle.39 It can be seen that the initial coulombic efficiency of PCM-2800 is higher than that of GPCM-900, as well as shorter discharge plateau at about 0.70 V, probably because HC (generated from sucrose) heated at 2800 °C can reduce irreversible capacity and form consolidated interface between active materials and electrolyte.40,41 In addition, the initial coulombic efficiencies of the non-graphitized (PCM-900) and the graphitized PCMs (PCM-2800 and GPCM-900) are higher than those of NC and GNC, respectively, suggesting the spherical morphology and amorphous carbon on the surface of an electrode (see above TEM images) can suppress the electrolyte decomposition remarkably and contribute to the formation of the stable SEI films.9–12,14 The observation of the discharge and charge plateaus below 0.20 V suggests the insertion/extraction of lithium ions from carbon for GNC, PCM-2800, GPCM-900, and GMs.18,42 Meanwhile, the charge and discharge curves for NC and PCM-900 are different from those of GNC, PCM-2800 and GPCM-900. For NC and PCM-900, no obvious plateaus can be found and it reveals there are no phase transition during the charge and discharge processes, while distinct voltage plateaus can be clearly identified below 0.20 V for GNC, PCM-2800 and GPCM-900, indicating that the different lithium ion storage mechanisms for different carbon structures.1,4,43
 | ||
| Fig. 5 Electrochemical properties of all the samples: (a) the initial discharge–charge curves at 50 mA g−1, (b) cyclic voltammograms during the initial discharge–charge, (c) cycling performance and corresponding coulombic efficiency at 50 mA g−1, (d) rate performance at different current densities, and (e) electrochemical impedance spectrum. | ||
| Sample | DC1sta | CC1stb | CE1stc | CC100thd | FRe | RC50f | RC100g | RC500h | RC1000i |
|---|---|---|---|---|---|---|---|---|---|
| a DC1st, (mA h g−1), the first discharge capacity.b CC1st, (mA h g−1), the first charge capacity.c CE1st, (%), the initial coulombic efficiency.d CC100th (mA h g−1), the 100th charge capacity.e FR, (%/cycle), fading rate per cycle, fading rate (%/cycle) = [(CC100th − CC1st)/CC1st]/100 × 100%.f RC50.g RC100.h RC500.i RC1000, (mA h g−1), average charge capacities at current densities of 50, 100, 500, and 1000 mA g−1, respectively. | |||||||||
| NC | 477 | 241 | 50.5 | 231 | −0.04 | 245 | 230 | 160 | 123 |
| GNC | 433 | 332 | 76.7 | 357 | +0.07 | 345 | 337 | 229 | 76 |
| PCM-900 | 475 | 266 | 56.0 | 263 | −0.01 | 263 | 257 | 209 | 149 |
| PCM-2800 | 394 | 351 | 89.2 | 347 | −0.01 | 349 | 344 | 315 | 160 |
| GPCM-900 | 416 | 338 | 81.4 | 348 | +0.03 | 337 | 332 | 299 | 156 |
| GMs | 412 | 354 | 85.8 | 344 | −0.03 | 356 | 326 | 191 | 90 |
Cyclic voltammograms in Fig. 5b reveal that a distinct cathodic peak attributed to the decomposition of electrolyte and the formation of SEI film at 0.6 V can be observed during the first sweep for all samples.12,14 Only a very small cathodic peak is observed for PCMs, which indicates that amorphous carbon on the surface of an electrode decrease the contact areas between GNC and electrolyte and can effectively suppress solvent decomposition on the graphite electrode. The peaks observed at around 0.1 and 0.01 V are assignable to the lithiation process of graphite.12 It can be seen that the oxidation peaks of PCMs (PCM-2800 and GPCM-900) are shifted toward lower potentials, compared to GMs, indicating that PCMs easily undergoes the electrochemical oxidation by accepting electrons during first lithium deintercalation.18,44 This is because the porous structure, small grain size, and the HC conductive network in PCMs could provide shorter and faster transport pathways for both electrons and Li ions. For NC and PCM-900, no obvious peaks can be found because there are no phase transition during the charge and discharge processes, indicating that the different lithium ion storage mechanisms for different carbon structures, consisted with Fig. 5a.
The cycling performance in Fig. 5c shows that the charge capacities of NC, PCM-900, GNC, PCM-2800, GPCM-900 and GMs are around 241, 266, 332, 351, 338, and 354 mA h g−1 at the first cycle, and 231, 263, 357, 347, 348 and 344 mA h g−1 after 100 cycles. Thus, their fading rates in Table 2 are all less than −0.1% during 100 cycles, indicating their stable cycling performance. Compared to NC and PCM-900, the higher capacities for GNC, PCM-2800, and GPCM-900 stems from their increased graphitization.4 It can be seen that NC and PCM-900 with low reversible capacities and stable cycling properties are different from other amorphous carbon with high capacities and fast fading on cycling. This is because NC powders are obtained at 1000 °C in industry, leading to significantly reducing the number of “cavities” that can insert large amounts of lithium and increase side reaction and hydrogen atoms, which play a crucial role in the mechanism of lithium insertion.45 The capacity of GNC is higher than that of PCM-2800 and GPCM-900, probably because of the presence of HC in the latter which have less contribution for capacity and more for weight. It is found that the capacities of graphitized microspheres (PCM-2800 and GPCM-900) are in the range of 340–350 mA h g−1 at the current density of 50 mA g−1 after 100 cycles, comparable to that of GMs (344 mA h g−1).
The rate performances of all the samples at different current densities between 0.005 and 2.0 V are shown in Fig. 5d. At low current densities (50 and 100 mA g−1), the observed results are consistent with that obtained in the above cycling test. However, the distinct difference in electrochemical properties is observed for these samples when the current density is increased to 500 and 1000 mA g−1. The average charge capacities of NC, GNC, PCM-900, PCM-2800, GPCM-900 and GMs in Table 2 are 160, 229, 209, 315, 299 and 191 mA h g−1 at 500 mA g−1, and 123, 76, 149, 160, 156, and 90 mA h g−1 at 1000 mA g−1, respectively. PCMs (PCM-900, PCM-2800 and GPCM-900) have much higher capacity than that of NC and GNC at the current density of 500 and 1000 mA g−1, because the porous structure, small grain size, and the HC conductive network in PCMs could provide shorter and faster transport pathways for both electrons and Li ions,46,47 and spherical structure allows easy diffusion of Li ions and electrochemically accessible through physical contact well with each other,48 thus leading to the higher rate capability. In addition, conductive carbon network plays a more important role in the kinetics performances and electrochemical properties along with increasing current density. The capacity of non-graphitized PCMs (PCM-900) is as high as 150 mA h g−1 even at 1000 mA g−1, indicating that non-graphitized PCMs is a promising material for the quick charge–discharge energy storage.
Fig. 5e shows the electrochemical impedance spectrum before cycling of all the samples. The impedance plot is consisted of one semicircle curve that corresponds to the resistance of the Li ion transfer through SEI layers in the high frequency range, and a straight line to the charge transfer resistance at the electrode–electrolyte interface and the Li ion Warburg diffusion resistance in the solid electrode material in the low frequency range.15 Although the slopes of their straight lines are very similar, the size of their semicircles is different, going as GMs < PCM-2800 < GPCM-900 < PCM-900 < GNC < NC. It should be pointed out that the PCMs (PCM-900, PCM-2800, and GPCM-900) show much smaller semicircle than those of NC and GNC, indicating a lower electrochemical reaction resistance in the PCMs electrode, probably because of the presence of the porous structure, small grain size, and the HC conductive network in PCMs.46 It can be seen that the electrochemical reaction resistances of GNC and PCM-2800 are lower than those of NC and PCM-900, respectively. It is considered that the electronic conductivity of carbon could be improved after graphitization (2800 °C). Although the GMs has the lowest resistance, its rate property is worse than that of PCMs. We think that it is influenced by structure in the microspheres. It is concluded that for the prepared PCMs, pore structure and the HC conductive network can improve the rate performance.22
Fig. 6 shows the top-view and cross-view SEM images of the electrode disks of PCM-2800 before and after 100 discharge–charge cycles at 50 mA g−1. The surface morphology of the electrode disks of fresh and cycled PCM-2800 in Fig. 6a and c shows that the original texture of electrode can be well retained in terms of shape, size, and structural integrity, indicating its good stability. The cross-sectional SEM images of fresh and cycled PCM-2800 in Fig. 6b and d show PCM-2800 microspheres are homogeneously dispersed within the anode mixed composites and spherical morphology can be well maintained after 100 cycles. It could be concluded that the structure of synthetic PCMs via the spray drying method is stable during lithium ion repeated intercalation/de-intercalation process, contributing to excellent electrochemical performance.
 | ||
| Fig. 6 The top-view and cross-view SEM images of the electrode disks of PCM-2800 before (a and b) and after (c and d) 100 discharge–charge cycles at 50 mA g−1. | ||
Although PCMs with excellent rate performance can be obtained via spray drying method, it should be noted that the tap density of PCMs (0.47–0.56 g mL−1) is less than that of GMs (1.18 g mL−1) because of the existence of inner porosity, leading to relatively low volume energy density of PCMs. It should be mentioned that these PCMs can be used in energy storage batteries for electrical network peak shaving, microgrid and off-grid wind power system.49,50 On the one hand, to improve the cycling stability of PCMs, surface modification, such as surface coating, will be further acted and could decrease surface porosity which contributes to form consolidated interface between active materials and electrolyte, and effective use inner porosity which benefits to reduce volume expansion of the electrodes.47 On the other hand, to improve the energy density of PCMs, some active materials with the higher capacity, such as Si nanoparticles51 and transition metal oxide nanoparticles,52 could be filled into the inner porous space of PCMs. Such work is underway.
4. Conclusions
We have employed the industrially established spray drying method for scalable synthesis of porous carbon microspheres (PCMs) from the waste fine carbon powders using sucrose as the binder. Similar to the commercial graphite, the obtained PCMs have a spherical morphology with an average particle size of 10–30 μm. After the heat treatment, the PCMs have developed the porous structure and hard carbon (HC) network. When used as anode materials for lithium-ion batteries, the measured charge capacities of PCM-2800 are 315 and 160 mA h g−1 at 500 and 1000 mA g−1, respectively, superior to that of GMs (191 and 90 mA h g−1). The initial coulombic efficiency of the graphitized PCM-2800 is 89.2%, higher than that of GMs (85.8%) and GPCM-900 (81.4%), indicating that amorphous HC on the surface of materials can suppress the electrolyte decomposition remarkably and contribute to forming stable SEI films. In addition, after 100 cycles, spherical morphology of PCMs can be well maintained, contributing to excellent electrochemical performance. The concept of using the spray granulation method can be applied to reuse the waste fine non-graphitized and graphitized carbon particles generated from the process of industrial production.Acknowledgements
The authors gratefully acknowledge the supports from the National Natural Science Foundation of China (no. 51272252, 51402302, 51402299) and Hundred Talents Program of the Chinese Academy of Sciences.References
- M. Endo, C. Kim, K. Nishimura, T. Fujino and K. Miyashita, Carbon, 2000, 38, 183–197 CrossRef CAS.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef CAS PubMed.
- M. Wissler, J. Power Sources, 2006, 156, 142–150 CrossRef CAS PubMed.
- S. Flandrois and B. Simon, Carbon, 1999, 37, 165–180 CrossRef CAS.
- Y. P. Wu, E. Rahm and R. Holze, J. Power Sources, 2003, 114, 228–236 CrossRef CAS.
- J. Chen, C. Yao, C. Cheng, W. Hurng and T. Kao, J. Power Sources, 1995, 54, 494–495 CrossRef CAS.
- S. Ma, J. Li, X. Jing and F. Wang, Solid State Ionics, 1996, 86–88, 911–917 CrossRef CAS.
- J. Shim and K. A. Striebel, J. Power Sources, 2007, 164, 862–867 CrossRef CAS PubMed.
- H. L. Zhang, S. H. Liu, F. Li, S. Bai, C. Liu, J. Tan and H. M. Cheng, Carbon, 2006, 44, 2212–2218 CrossRef CAS PubMed.
- M. Yoshio, H. Wang and K. Fukuda, Angew. Chem., Int. Ed., 2003, 115, 4335–4338 CrossRef.
- M. Yoshio, H. Wang, K. Fukuda, Y. Hara and Y. Adachi, J. Electrochem. Soc., 2000, 147, 1245–1250 CrossRef CAS PubMed.
- C. Wang, H. Zhao, J. Wang, J. Wang and P. Lv, Ionics, 2012, 19, 221–226 CrossRef.
- B. Veeraraghavan, J. Paul, B. Haran and B. Popov, J. Power Sources, 2002, 109, 377–387 CrossRef CAS.
- H. Y. Lee, J. K. Baek, S. W. Jang, S. M. Lee, S. T. Hong, K. Y. Lee and M. H. Kim, J. Power Sources, 2001, 101, 206–212 CrossRef CAS.
- J. Yu, H. Zhan, Y. Wang, Z. Zhang, H. Chen, H. Li, Z. Zhong and F. Su, J. Power Sources, 2013, 228, 112–119 CrossRef CAS PubMed.
- X. Zhu, H. Chen, Y. Wang, L. Xia, Q. Tan, H. Li, Z. Zhong, F. Su and X. Zhao, J. Mater. Chem. A, 2013, 1, 4483–4489 CAS.
- C. Ma, Y. Zhao, J. Li, Y. Song, J. Shi, Q. Guo and L. Liu, Electrochim. Acta, 2013, 112, 394–402 CrossRef CAS PubMed.
- C. Ma, Y. Zhao, J. Li, Y. Song, J. Shi, Q. Guo and L. Liu, Carbon, 2013, 64, 553–556 CrossRef CAS PubMed.
- F. Su, X. Zhao, Y. Wang and J. Y. Lee, Microporous Mesoporous Mater., 2007, 98, 323–329 CrossRef CAS PubMed.
- Z. J. Fan, J. Yan, T. Wei, G. Q. Ning, L. J. Zhi, J. C. Liu, D. X. Cao, G. L. Wang and F. Wei, ACS Nano, 2011, 5, 2787–2794 CrossRef CAS PubMed.
- Y. Fang, Y. Lv, R. Che, H. Wu, X. Zhang, D. Gu, G. Zheng and D. Zhao, J. Am. Chem. Soc., 2013, 135, 1524–1530 CrossRef CAS PubMed.
- L. Zhang, M. Zhang, Y. Wang, Z. Zhang, G. Kan, C. Wang, Z. Zhong and F. Su, J. Mater. Chem. A, 2014, 2, 10161–10168 CAS.
- B. S. Lee, S. B. Son, K. M. Park, G. Lee, K. H. Oh, S. H. Lee and W. R. Yu, ACS Appl. Mater. Interfaces, 2012, 4, 6702–6710 CAS.
- B. S. Lee, J. H. Seo, S. B. Son, S. C. Kim, I. S. Choi, J. P. Ahn, K. H. Oh, S. H. Lee and W. R. Yu, ACS Nano, 2013, 7, 5801–5807 CrossRef CAS PubMed.
- E. Unur, S. Brutti, S. Panero and B. Scrosati, Microporous Mesoporous Mater., 2013, 174, 25–33 CrossRef CAS PubMed.
- K. Wenelska, K. Kierzek, R. J. Kalenczuk, X. Chen and E. Mijowska, ACS Appl. Mater. Interfaces, 2013, 5, 3042–3047 CAS.
- R. Vehring, Pharm. Res., 2008, 25, 999–1022 CrossRef CAS PubMed.
- C. Boissiere, D. Grosso, A. Chaumonnot, L. Nicole and C. Sanchez, Adv. Mater., 2011, 23, 599–623 CrossRef CAS PubMed.
- J. Meeus, D. J. Scurr, K. Amssoms, M. C. Davies, C. J. Roberts and G. Van den Mooter, Mol. Pharmacol., 2013, 10, 3213–3224 CrossRef CAS PubMed.
- W. S. Cheow, S. Li and K. Hadinoto, Chem. Eng. Res. Des., 2010, 88, 673–685 CrossRef CAS PubMed.
- S. G. Maas, G. Schaldach, E. M. Littringer, A. Mescher, U. J. Griesser, D. E. Braun, P. E. Walzel and N. A. Urbanetz, Powder Technol., 2011, 213, 27–35 CrossRef CAS PubMed.
- A. Carné-Sánchez, I. Imaz, M. Cano-Sarabia and D. Maspoch, Nat. Chem., 2013, 5, 203–211 CrossRef PubMed.
- D. S. Jung, T. H. Hwang, S. B. Park and J. W. Choi, Nano Lett., 2013, 13, 2092–2097 CrossRef CAS PubMed.
- J. Ying, C. Wan, C. Jiang and Y. Li, J. Power Sources, 2001, 99, 78–84 CrossRef CAS.
- D. Bom, R. Andrews, D. Jacques, J. Anthony, B. Chen, M. S. Meier and J. P. Selegue, Nano Lett., 2002, 2, 615–619 CrossRef CAS.
- Y. Wang, F. Su, C. D. Wood, J. Y. Lee and X. S. Zhao, Ind. Eng. Chem. Res., 2008, 47, 2294–2300 CrossRef CAS.
- L. Xie, H. Wang, C. Jin, X. Wang, L. Jiao, K. Suenaga and H. Dai, J. Am. Chem. Soc., 2011, 133, 10394–10397 CrossRef CAS PubMed.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cancado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276–1291 RSC.
- A. J. Smith, J. C. Burns, X. Zhao, D. Xiong and J. R. Dahn, J. Electrochem. Soc., 2011, 158, A447–A452 CrossRef CAS PubMed.
- E. Buiel, A. George and J. Dahn, J. Electrochem. Soc., 1998, 145, 2252–2257 CrossRef CAS PubMed.
- E. Buiel and J. Dahn, Electrochim. Acta, 1999, 45, 121–130 CrossRef CAS.
- L. Z. Bai, D. L. Zhao, T. M. Zhang, W. G. Xie, J. M. Zhang and Z. M. Shen, Electrochim. Acta, 2013, 107, 555–561 CrossRef CAS PubMed.
- K. Sato, M. Noguchi, A. Demachi, N. Oki and M. Endo, Science, 1994, 264, 556–558 CAS.
- I. A. Profatilova, S. S. Kim and N. S. Choi, Electrochim. Acta, 2009, 54, 4445–4450 CrossRef CAS PubMed.
- J. Dahn, T. Zheng, Y. Liu and J. Xue, Science, 1995, 270, 590–593 CAS.
- S. Xin, Y. G. Guo and L. J. Wan, Acc. Chem. Res., 2012, 45, 1759–1769 CrossRef CAS PubMed.
- T. H. Park, J. S. Yeo, M. H. Seo, J. Miyawaki, I. Mochida and S. H. Yoon, Electrochim. Acta, 2013, 93, 236–240 CrossRef CAS PubMed.
- T. Wei, F. Wang, J. Yan, J. Cheng, Z. Fan and H. Song, J. Electroanal. Chem., 2011, 653, 45–49 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- I. Hadjipaschalis, A. Poullikkas and V. Efthimiou, Renewable Sustainable Energy Rev., 2009, 13, 1513–1522 CrossRef PubMed.
- U. Kasavajjula, C. Wang and A. J. Appleby, J. Power Sources, 2007, 163, 1003–1039 CrossRef CAS PubMed.
- M. Reddy, G. Subba Rao and B. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |