DOI:
10.1039/C4RA15234D
(Paper)
RSC Adv., 2015,
5, 17953-17966
Functional tuning of organic dyes containing 2,7-carbazole and other electron-rich segments in the conjugation pathway†
Received
25th November 2014
, Accepted 4th February 2015
First published on 4th February 2015
Abstract
Organic dyes containing a triarylamine donor, a cyanoacrylic acid acceptor and a conjugation pathway composed of 2,7-carbazole, thiophene and fluorene have been synthesized and characterized as sensitizers for TiO2-based dye-sensitized solar cells. The effect of the nature of the conjugation bridge on the optical, electrochemical and photovoltaic properties has been investigated. Elongation of conjugation by the insertion of 2,7-carbazole or 2,7-fluorene π-linkers led to an increase in the molar extinction coefficients while the use of terthiophene red-shifted the absorption. Also the lengthening of the π-bridge helped to raise the lowest unoccupied molecular orbital of the dyes relative to the conduction band of TiO2. This increased the thermodynamic driving force for the electron injection from the excited dyes into the conduction band of TiO2. The trends in the optical properties of the dyes were also substantiated by TDDFT computations. DSSCs based on a dye containing terthiophene in the conjugation bridge exhibited the highest power conversion efficiency (4.9%) in the series attributable to its higher photocurrent (JSC = 13.08 mA cm−2) generation. However, the dye with a fluorene linker exhibited high open circuit voltage (VOC = 632 mV) and prolonged electron lifetime for the device. The role of the π-bridge in the charge transfer and electron recombination kinetics in the DSSC was elucidated by electrochemical impedance spectroscopic studies.
Introduction
Organic dyes featuring the donor–linker–acceptor (D–π–A) configuration have emerged as excellent candidates for the conversion of solar light into electricity and rival ruthenium-based inorganic dyes.1,2 The limited availability, toxic nature and tedious synthetic & purification protocols of ruthenium-based dyes hamper their wide potential towards applications.1,2 While for organic dyes, flexibility in design, higher molar extinction coefficients and cheap synthetic methods make them better alternatives.1 Even though organic dyes are more intensely absorbing they achieved lower efficiency compared to metal containing dyes and perovskite solar cells.2,3 Aggregation is one of the major drawbacks of the organic dye-sensitized solar cells (DSSCs). It has been observed that the triarylamine units which have twisted arrangement greatly benefit the structural requirements to retard the aggregation at the surface of TiO2 besides sufficing the required donor strength to impart effective charge migration.4 The strength of donor–acceptor interactions plays a vital role in photo induced intra-molecular charge transfer (ICT) from donor to acceptor. Replacement of phenyl groups in the triphenylamine unit with heterocyclic electron-rich fragments though increase the donor strength often led to mismatch in the highest occupied molecular orbital (HOMO) energy levels with the electrolyte and sluggish the dye regeneration.5 So identifying chromophores which retain the dye regeneration capabilities but alter the excited state characteristics suitably is an important task.
Development of potential structural fragments for longer wavelength absorption is an additional requirement to achieve effective light harvesting in the visible region. Introduction of electron-rich linkers has been found to fine-tune the absorption properties in the longer wavelength region.6 Dyes composed of planar aromatic linker assist the interactions between the donor and acceptor units.7 It was found that the introduction of fluorene and carbazole benefits the optical and electrochemical properties.8 Fluorene units generally increase the molar extinction coefficients in the absorption and impart redox stability to the molecule.5c,8a–c Similarly, carbazole has good hole transport property,9 and found to be an effective conjugating segment in our earlier study.8e,f However, elongated planarized conjugation of linker suffers from aggregation which diminishes device efficiency.10 The peripheral or lateral alkyl groups and twisted geometry of the dye diminish the aggregation on TiO2 surface.11 Recently, Bäuerle et al. reported that the back electron transfer can be efficiently blocked by introducing a twisted phenyl spacer and demonstrated a 6.5 times hike in efficiency.11g
In this work, we have synthesized five new dyes by incorporating the various aromatic π-linkers in between the diphenylaminocarbazole donor and cyanoacrylic acid acceptor. The linkers were chosen to exploit the different beneficial properties reported in the literature for such units.12 We have used phenyl, terthiophene, fluorene and carbazole linkers to design the new dyes. We find that the use of terthiophene led to red-shifted absorption while the tilting in phenyl conjugated dye and elongation of conjugation in fluorene and carbazole containing dyes produced relatively blue-shifted absorption profiles. Since the electron richness of these conjugating segments is almost similar the differences in the electronic properties mainly originated from the lowest unoccupied molecular orbital (LUMO) alternations. This led to profound effect on the electron injection kinetics of the excited dye into the conduction band of TiO2 and the overall efficiency of the DSSC.
Results and discussions
Synthesis and characterization
The synthetic scheme used to prepare the target dyes is shown in Scheme 1 and the structure of the dyes in Fig. 1. In the first step, 9-butyl-N,N-diphenyl-7-(thiophen-2-yl)-9H-carbazol-2-amine (2) was prepared by performing Stille coupling13 reaction of 7-bromo-9-butyl-N,N-diphenyl-9H-carbazol-2-amine8e (1) with tributyl(thiophen-2-yl)stannane using PdCl2(PPh3)2 as catalyst in DMF. Later 2 was treated with n-butyl lithium followed by quenching with tri-n-butyl-tin chloride gave the corresponding tin reagent. This tin reagent was used as a coupling partner with several bromoaryl(heteroaryl)carboxaldehydes8f,14 to obtain the desired aldehyde derivatives (3a–e). Finally, the target dyes (4a–e) were obtained in good yields by treating with cyanoacetic acid under standard Knoevenagel condensation15 conditions. The dyes are brown to black in color and soluble in common organic solvents such as cyclohexane (CH), toluene (Tol), dichloromethane (DCM), tetrahydrofuran (THF), N,N-dimethylformamide (DMF) and acetonitrile (ACN). All the compounds were thoroughly characterized by IR, NMR (1H and 13C) and mass spectral methods.
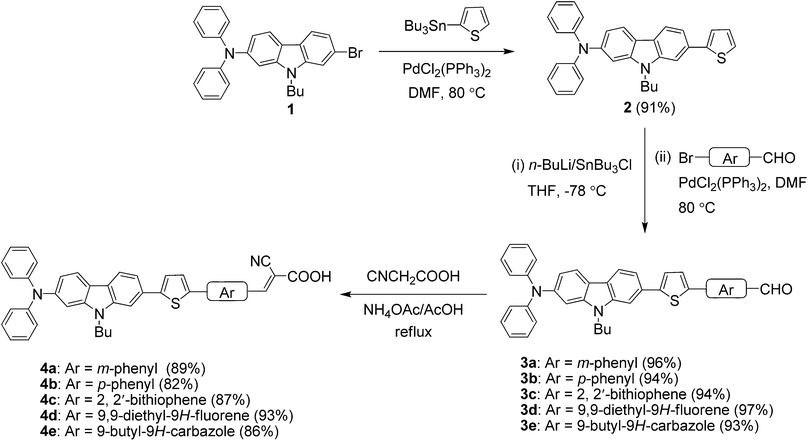 |
| | Scheme 1 Synthetic scheme to prepare target dyes. | |
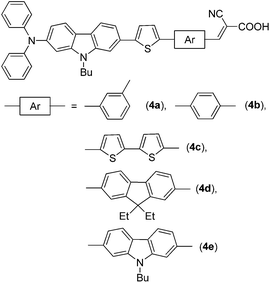 |
| | Fig. 1 Structures of the carbazole-based dyes containing extended π-conjugation. | |
Photophysical properties
The absorption spectra of the dyes recorded in dichloromethane solution are displayed in Fig. 3 and the relevant data compiled in Table 1. All the dyes, except 4a, exhibit at least three distinguishable absorption peaks. The shorter wavelength absorption peak at ca. 275 nm is attributed to the carbazole localized π–π* transition.8e,16 This assignment is further supported by the fact that the intensity of this absorption is high for the dye 4e which contains an additional carbazole in the conjugation pathway. A weak absorption occurring at ∼310 nm is originating probably from the n–π* transition. The longer wavelength absorption at ∼390 for all the dyes is due to the π–π* transition originating from the orbitals delocalized over the entire conjugation pathway. The furthest absorption peak is assigned as a transition originating from the charge transfer (CT) from the triarylamine donor to the cyanoacrylic acid acceptor.8e The peak wavelengths of the CT transition in the dyes assume the order: 4a (391 nm) < 4d (420 nm) < 4e (439 nm) < 4b (448 nm) < 4c (498 nm) which is reminiscent of the alternations in the conjugation pathway. The dyes (4b and 4c) with the shortest conjugation in the linker displayed relatively red-shifted absorption than the dyes (4d and 4e) with longest conjugation pathway. Significantly blue-shifted absorption realized for the dye 4a, possessing the non-conjugated donor and acceptor (meta linkage), is self-explanatory. The carbazole-containing dye, 4e, exhibited broad and intense absorption when compared to the fluorene conjugated dye, 4d. This is partially due to the electron-richness of carbazole which resulted in the slight red-shift (Δλ = 19 nm) in the CT transition. Also the π–π* transition is marginally blue-shifted for the phenylene-conjugated dye (4b) than those observed for the other dyes 4c, 4d and 4e. This apparently points that the conjugation length is almost same in all the latter dyes or have attained the conjugation saturation.
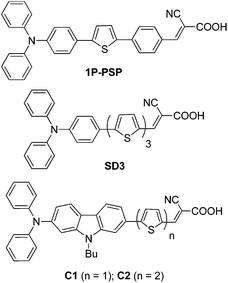 |
| | Fig. 2 Structures of the related dyes. | |
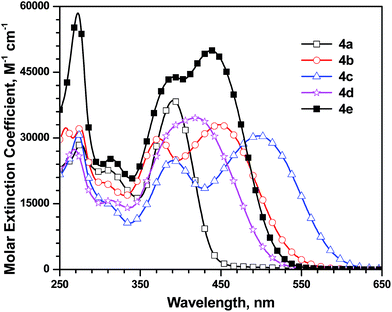 |
| | Fig. 3 Absorption spectra of the dyes recorded in dichloromethane. | |
Table 1 Optical and electrochemical properties of the aldehydes and dyes recorded in dichloromethane solutionsa
| Compd |
λabs, nm (εmax × 103 M−1 cm−1) |
λem, nm |
Stokes shiftb, cm−1 |
Eoxc, V |
HOMOd, eV |
LUMOe, eV |
E0–0f, eV |
E*oxg, V |
na = not applicable. From 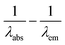 . Oxidation potentials are reported with reference to the ferrocene internal standard. Deduced from the oxidation potential using the formula HOMO = 4.8 + Eox. Deduced using the formula LUMO = HOMO − E0–0. Calculated from intersection of absorption and emission spectra. Calculated from E*ox = Eox − E0–0, with respect to NHE. Reported for THF solutions. . Oxidation potentials are reported with reference to the ferrocene internal standard. Deduced from the oxidation potential using the formula HOMO = 4.8 + Eox. Deduced using the formula LUMO = HOMO − E0–0. Calculated from intersection of absorption and emission spectra. Calculated from E*ox = Eox − E0–0, with respect to NHE. Reported for THF solutions. |
| PSPh |
427 (29.0) |
na |
na |
0.79 |
5.29 |
2.77 |
2.53 |
−1.73 |
| SD3h |
480 (46.2), 386 (22.4), 302 (20.7) |
na |
na |
0.43 (84), 0.57 (65) |
5.23 |
3.06 |
2.17 |
−0.97 |
| C1 |
472 (29.3), 357 (18.0), 311 (16.8) |
572 |
3704 |
0.38 (65), 0.86 (53) |
5.18 |
2.85 |
2.33 |
−1.18 |
| C2 |
489 (34.3), 375 (23.5), 311 (16.9), 264 (34.6) |
579 |
3179 |
0.33 (52), 0.71 (73) |
5.13 |
2.86 |
2.27 |
−1.17 |
| 2 |
375 (30.1), 313 (17.3), 274 (36.8) |
432 |
3519 |
0.33 (72), 0.81 |
5.13 |
2.04 |
3.09 |
−1.99 |
| 3a |
393 (36.1), 317 (18.9), 271 (28.2) |
486 |
4869 |
0.33 (69), 0.74 |
5.13 |
2.31 |
2.82 |
−1.72 |
| 3b |
410 (30.2), 325 (15.9), 272 (24.4) |
569 |
6816 |
0.34 (73), 0.77 |
5.14 |
2.59 |
2.55 |
−1.44 |
| 3c |
441 (32.6), 308 (14.6), 267 (31.4) |
565 |
4977 |
0.32 (75), 0.61 |
5.12 |
2.67 |
2.45 |
−1.36 |
| 3d |
411 (39.4), 308 (18.6), 258 (29.6) |
544 |
5949 |
0.32 (65), 0.64 |
5.12 |
2.52 |
2.60 |
−1.51 |
| 3e |
410 (55.2), 261 (55.8) |
524 |
5306 |
0.32 (66), 0.61 |
5.12 |
2.48 |
2.64 |
−1.55 |
| 4a |
391 (38.6), 311 (22.6), 276 (28.6) |
481 |
5456 |
0.32 (63), 0.72 |
5.12 |
2.29 |
2.83 |
−1.74 |
| 4b |
448 (33.0), 374 (29.8), 276 (32.2) |
500 |
2321 |
0.32 (62), 0.72 |
5.12 |
2.53 |
2.59 |
−1.50 |
| 4c |
498 (30.5), 391 (24.8), 273 (30.7) |
565 |
2381 |
0.30 (63), 0.62 |
5.10 |
2.77 |
2.33 |
−1.26 |
| 4d |
420 (34.6), 314 (15.7), 268 (27.4) |
513 |
4373 |
0.30 (63), 0.64 |
5.10 |
2.51 |
2.59 |
−1.52 |
| 4e |
439 (50.0), 391 (43.9), 314 (25.2), 273 (58.4) |
526 |
3768 |
0.30 (61), 0.60 |
5.10 |
2.55 |
2.55 |
−1.45 |
The important outcome of the extension of conjugation in the present dyes is the improvement in the molar extinction coefficients for the CT transition. All the dyes exhibited high molar extinction coefficients when compared to the simple dyes (C1 and C2; Fig. 2) containing thiophene or bithiophene in the conjugation.8e Also, it is interesting to compare the dyes 4b and 4c with those dyes (1P-PSP (ref. 12d) and SD3 (ref. 8b); Fig. 2) containing phenyl unit in place of carbazole. The dyes 4b and 4c exhibited longer wavelength CT transition (Δλ ≥ 18 nm) when compared to the phenyl analogues (1P-PSP (ref. 12d) and SD3 (ref. 8b)). This clearly highlights the beneficial role of 2,7-disubstituted carbazole as a linker in improving the absorption characteristics of the dyes.
We have also measured the absorption spectra of the dyes in solvents of different polarity to ascertain the strength of CT in these molecules. The absorption spectra of all the dyes with the exception of 4a exhibited a negative solvatochromism for the longer wavelength electronic transition as illustrated in the representative example displayed in Fig. 4. This clearly confirms that the dyes possess strong intramolecular donor–acceptor interaction in the ground state which results in a polar ground state structure due to the migration of charge density from the amine donor to the cyanoacrylic acid acceptor.
 |
| | Fig. 4 Absorption spectra of the dye 4c recorded in different solvents. | |
Generally, the polar molecules are effectively solvated by the polar solvents and relaxed in energy.17 On the contrary the non-polar solvents cannot have a stabilizing interaction with the polar dye molecules.17 Consequently, the dyes exhibit red-shifted absorption in nonpolar solvents and blue-shifted λmax in polar solvents. The dyes displayed red-shifted absorption in non-polar solvents such as toluene. The most red-shifted absorption observed for dichloromethane solutions arise due to the instant stabilization of the polarizable electrons and is commonly observed for the cationic dyes in chloro-solvents.18 Similarly, the hypsochromic effect observed for the dyes in THF and DMF solutions may be attributed to the hydrogen bonding between carboxylic acid unit of the dyes and THF and basic nature of DMF deprotonate the carboxylic acid, respectively.7a,12c Absence of solvatochromic response for 4a is self-explanatory as it lacks donor–acceptor interactions due to meta-linkage.19
Further we have confirmed the presence of ICT transition in the dyes by the addition of the trifluoroacetic acid (TFA) and triethylamine (TEA) to the dye solutions in dichloromethane. The addition of acid/base to the dyes affects the acid–base equilibrium to shift towards neutral or deprotonated states. The response of 4d on addition of TFA/TEA is displayed in Fig. 5 as an illustration. Again, the dye 4a was inert to the addition of TFA/TEA. A slight bathochromic shift on addition of TFA and significant hypsochromic shift with TEA for the longer wavelength absorption in these molecules are suggestive of its ICT origin.20
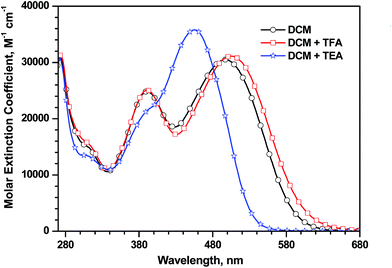 |
| | Fig. 5 Absorption spectra of the dye 4c recorded in dichloromethane, after the addition of TFA and TEA. | |
Absorption spectra recorded for the dyes anchored on TiO2 films are displayed in Fig. 6. The dye 4c exhibited most red-tailing in the absorption spreading upto 650 nm. In general, all dyes (4b–e) displayed a bathochromic shift (28–35 nm) when anchored on TiO2 film. The red-shift in the absorption may be rationalized by considering J-type aggregate formation between the dyes at the surface of TiO2.21 The dye 4d showed comparatively small red-shift, probably, due to the alkyl groups present at C9 of fluorene which may impose hindrance for the close assembly of the dyes when compared to its congener dye 4e which contains carbazole bearing one alkyl chain.22
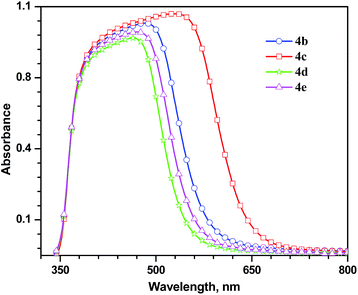 |
| | Fig. 6 Absorption spectra of the dyes (4b–e) recorded for TiO2 films. | |
All the dyes are weakly emitting in dichloromethane solutions with blue to yellow emission as displayed in Fig. 7. The dyes 4b and 4c exhibited broad asymmetric emission profiles compared to the rest. This probably indicates the presence of two different excited states, namely, locally excited (LE) and charge transfer (CT) states.23 In such compounds, the high energy band is assigned to the LE while the longer wavelength transition originates from the CT state. On this basis, it can be ascertained from the emission profiles that the LE state is more populated for 4b while CT state is predominant in 4c. It is safe to assume that the twisted phenyl linker in 4b disfavours the molecular excitation into the CT state. Similarly, for 4a, the meta linkage forbids the CT in the ground state and excited state realized in nonpolar solvents.24 However, the red-shifted broad emission observed for 4a in polar solvents such as ACN and DMF is indicative of CT.25 CT state population in the meta-conjugated donor–acceptor compounds have been attributed to the structural reorganization inducing necessary orbital overlap.24 Interestingly, 4a showed positive solvatochromism while the remaining dyes exhibited negative solvatochromism in the excited state. This clearly points that 4a is relatively stabilized in excited state by polar solvents while the other dyes are effectively solvated by the polar solvents in the ground state.
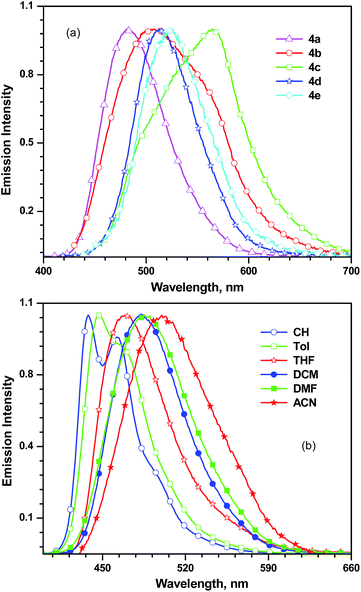 |
| | Fig. 7 Emission spectra of the dyes (a) 4a–e recorded in dichloromethane and (b) 4a recorded in different solvents. | |
The solvent dependent photophysical parameters of the dyes were analysed by Lippert–Mataga plot26 and Stokes shift vs. ET(30) parameter27 correlation. The dye 4a and 4d displayed linear variation in the plots as displayed in Fig. 8. This indicates that the dyes 4a and 4d features solvent effect in the excited state. For the dyes 4b, 4c and 4e these plots did not follow linear trend. This may be attributed to the presence of specific interactions such as hydrogen bonding, dipole–dipole interactions, etc.25a,b,28
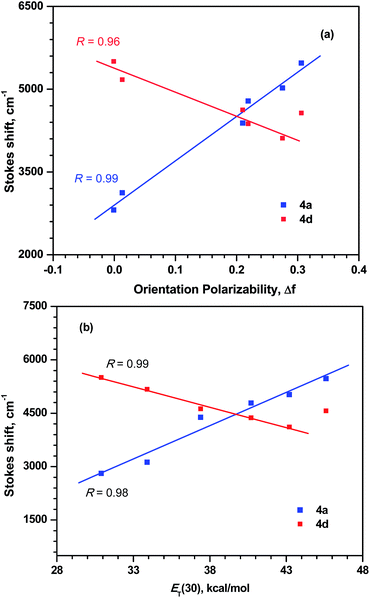 |
| | Fig. 8 (a) Lippert–Mataga and (b) Stokes shift vs. ET(30) parameter plots for the dyes 4a and 4d. | |
Electrochemical properties
The orbital energies of the dyes played a vital role in the electron injection and dye regeneration of the dye during the DSSC operation. In order to estimate these parameters, the redox behaviour of the dyes was analysed by using cyclic voltammetry (CV) in dichloromethane. All the dyes exhibited two oxidation processes. The quasi-reversible oxidation couple (shown in ESI†) occurring at low positive potentials is attributed to the removal of electron from the triarylamine unit while the irreversible oxidation at relatively high oxidation potentials originate from the oxidation of conjugation pathway. All the dyes possess similar first oxidation potentials (Table 1) indicative of negligible influence of structural variation in linkers on oxidation potential of the dyes. Interestingly, all the dyes showed low oxidation potentials than C1. There are two reasons for this. Firstly, elongation of conjugation reduces the interaction between the donor and acceptor.10 Secondly, the introduction of electron-rich segments such as fluorene, carbazole and terthiophene in the conjugation pathway facilitates the oxidation.7a,8e,12b,c
The ground and excited state redox potentials derived from the electrochemical parameters and optical band gap is compared with the conduction band (CB) of TiO2 and redox potential of the electrolyte (I−/I3−) in Fig. 9. All the dyes possess more positive ground state redox potentials (∼1.08 V vs. NHE), than the redox potential of the electrolyte (0.4 V vs. NHE).29 The difference, 0.68 V is sufficient enough to provide the required thermodynamic driving force for the regeneration of the dye by electrolyte.1 The excited state oxidation potential of the dyes ranged between −1.26 V to −1.74 V vs. NHE, which are more negative than CB of TiO2 (−0.5 V vs. NHE).29 These values suggest an energetically favourable electron injection from the excited dye molecules into the CB of TiO2. The dyes 4b, 4d and 4e possess excited state oxidation potentials close to ∼1.50 V but the dye 4c inherits lower value at −1.26 V. This probably may lead to lower electron injection rate for the dye 4c.
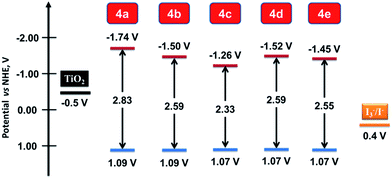 |
| | Fig. 9 Comparison of the ground and excited state redox potentials of the dyes (4a–e). | |
Theoretical calculations
To understand the nature of electronic transitions and the fragments contributing to the absorption we have performed density functional theory (DFT) computations on the dyes.30 The geometry of the dye was optimized first using B3LYP31 functional and 6-31G(d, p) basis set. The optimized structures were then used for TDDFT computations at MPW1K/6-31G(d, p)32 and BMK33/dgdzvp34 levels. The frontier molecular orbitals of the dyes 4b–e involved in the longer wavelength absorption are displayed in Fig. 10 along with the excitation energies obtained using different theoretical models. More details about the computational results are available in Table 2.
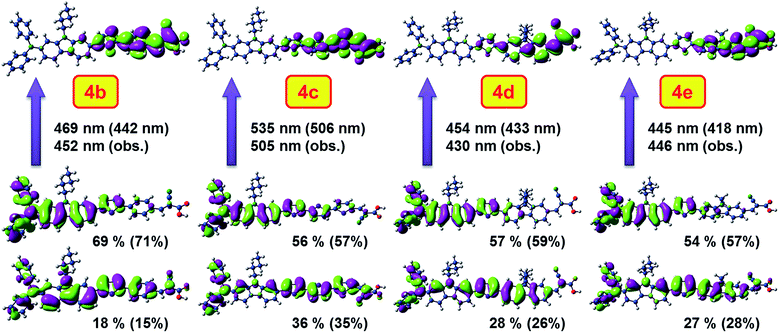 |
| | Fig. 10 Comparison of HOMO−2 or HOMO−1 (bottom), HOMO (middle) to LUMO (top) electronic excitation energies from BMK/dgdzvp and MPW1K/6-31G(d,p) (in parenthesis) computations (dye codes change). | |
Table 2 Computed vertical transition energies and their oscillator strengths, assignments, dipole moments and band gaps for the dyes by using MPW1K/6-31G(d,p) level for THF solvent
| Dye |
λ |
f |
Assignments |
μg, D |
HOMO, eV |
LUMO, eV |
E0–0, eV |
| 4a |
403 |
0.17 |
HOMO → LUMO (78%), HOMO−2 → LUMO (16%) |
8.03 |
−5.86 |
−2.13 |
3.73 |
| 4b |
442 |
1.66 |
HOMO → LUMO (69%), HOMO−2→LUMO (18%) |
7.51 |
−5.86 |
−2.29 |
3.57 |
| 4c |
506 |
2.07 |
HOMO → LUMO (56%), HOMO−1 → LUMO (36%) |
12.78 |
−5.83 |
−2.50 |
3.33 |
| 4d |
433 |
1.69 |
HOMO → LUMO (57%), HOMO−1 → LUMO (28%) |
6.46 |
−5.82 |
−2.22 |
3.60 |
| 4e |
418 |
1.92 |
HOMO → LUMO (54%), HOMO−1 → LUMO (27%) |
9.82 |
−5.82 |
−2.11 |
3.72 |
HOMO is mainly distributed over the diphenylaminocarbazole and thiophene units, while the LUMO mainly spread on the aromatic/heteroaromatic linker and cyanoacrylic acid segment. The prominent absorption occurring above 400 nm is mainly composed of electronic excitations from HOMO and HOMO−1 orbitals to the LUMO orbital in the dyes 4c–e. In the dye 4b, for this absorption besides HOMO to LUMO electronic excitation, a contribution from HOMO−2 to LUMO is also present. However, the organization of HOMO−2 in 4b is similar to that of HOMO−1 in 4c–e. It is a π-type orbital distributed over the entire aromatic conjugation in the molecules. It is to be noted here that out of these two contributions HOMO to LUMO excitation dominates with 50% share for this absorption. So this absorption can be mainly considered as charge transfer transition with minor contribution from the π–π* transition. All the dyes except 4a possess high oscillator strength for this absorption. The low oscillator strength observed for 4a is attributable to the delinking of HOMO and LUMO orbitals while the high values of 4c and 4e are reflective of the presence of electron-rich terthiophene and carbazole units, respectively in the conjugation pathway. Interestingly, the computations using MPW1K/6-31G(d,p)32 and PCM35 solvation model gave closely matching predictions for the dyes 4b–d. But for the dye 4e the computed parameters using BMK33/dgdzvp34 and SMD36 solvation model are more realistic. This clearly points that the results from the different functionals need to be treated with caution.
DSSC characteristics
To evaluate the light harvesting ability of the dyes (4b–e) they were used as sensitizers in nanocrystalline TiO2-based DSSC. The photovoltaic measurements were performed under AM 1.5 G simulated solar light at 100 mW cm−2 and the relevant data compiled in Table 3. The incident photon to current conversion efficiency spectrum (IPCE) and I–V characteristics of the devices are shown in Fig. 11. Among the dyes, 4c exhibits best photon to electron conversion characteristics with the peak value of 82% at 440 nm. The IPCE spectra of the devices are reminiscent of the absorption spectra of the corresponding dyes. Among the dyes, 4c showed promising device characteristics. The better power conversion efficiency of the device based on this device is attributed to the good light harvesting property of the dye which resulted in reasonably high photocurrent generation.
Table 3 Photovoltaic parameters of the dyes
| Dye |
η (%) |
VOC, mV |
JSC, mA cm−2 |
ff |
Rrec, ohm |
Rct2, ohm |
τe, ms |
| 4b |
3.05 ± 0.03 |
573.93 ± 2.01 |
8.18 ± 0.12 |
0.65 ± 0.01 |
31.97 |
33.17 |
1.16 |
| 4c |
4.84 ± 0.04 |
582.21 ± 0.67 |
13.00 ± 0.08 |
0.64 ± 0.00 |
39.90 |
23.38 |
3.12 |
| 4d |
3.11 ± 0.00 |
636.94 ± 4.18 |
7.32 ± 0.04 |
0.67 ± 0.00 |
42.81 |
37.78 |
8.41 |
| 4e |
2.44 ± 0.03 |
566.03 ± 1.15 |
7.68 ± 0.02 |
0.56 ± 0.01 |
30.34 |
34.58 |
1.16 |
| N719 |
7.84 ± 0.07 |
733.17 ± 5.75 |
16.53 ± 0.11 |
0.65 ± 0.01 |
— |
— |
— |
 |
| | Fig. 11 I–V Characteristics (a) and IPCE spectra (b) of the devices fabricated using the dyes 4b–e. | |
Electrochemical impedance spectroscopy
The interfacial charge transfer resistance greatly governs the efficiency of the DSSC. To scrutinize the effect of molecular structures on the electron transfer kinetics under dark and illumination conditions, we measured the electrochemical impedance spectroscopy (EIS) under forward bias in a frequency range from 10 mHz to 65 KHz. Fig. 12 shows Nyquist plots obtained for the devices under dark conditions. In principle, Nyquist plot shows three semicircles which corresponds the charge transfer resistances at Pt electrode/electrolyte and TiO2/dye/electrolyte interfaces and Warburg diffusion of I− in the electrolyte interface respectively. Radius of the larger semicircle in the Nyquist plot measured under dark condition corresponds to resistance of electron recombination (Rrec) TiO2/dye/electrolyte interface. Larger Rrec indicates the good inhibition for the recombination of electrons in CB of TiO2 with oxidized species (I3−) of electrolyte or dye.37 The Rrec recorded for the devices fabricated using the dyes assume the order, 4e < 4b < 4c < 4d. Since the donor and acceptor parts are similar in the dyes, the differences in Rrec can be ascribed to the nature of the linker. High Rrec observed for 4d is probably originating from the fluorene moiety.38 Reduction of dark current in DSSC may arise due to the inhibition of approach of electrolyte toward the TiO2 surface or energetically untenable situation for the injection of electron into the LUMO of the oxidized dye.1 Probably, the alkyl groups on C9 position of fluorene provides a hydrophobic environment to repel the ionic electrolyte from approaching TiO2 surface.8b,22,39
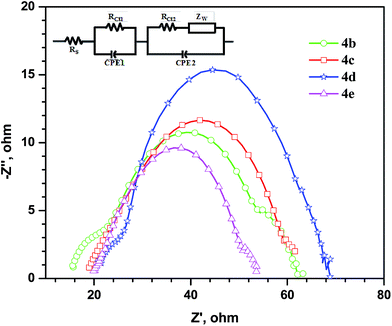 |
| | Fig. 12 Nyquist plots observed for the DSSCs measured under dark. Inset shows the equivalent circuit used to fit the EIS parameters. | |
Fig. 13 shows Nyquist and Bode phase plots of the devices under illumination. This information can be used to ascertain the charge transfer resistance (Rct2) at the interface of TiO2/dye/electrolyte and electron lifetime. The order of Rct2 in the devices is in the order, 4c < 4b < 4e < 4d with respect to the sensitizers. The lowest Rct2 was realized for the device containing 4c while the dye 4d exhibited the maximum Rct2 in the series. Electron injection into the CB of TiO2 from the dye is mainly dependent upon the charge separation in the molecular level and thermodynamic driving force.40 Since all the dyes possess required thermodynamic driving force (>0.3 V) the differences if any may originate due to variations in charge separation at the molecular level which may lead to sufficient dipole interactions at the TiO2/dye interface.1a,41 Probably, planar conjugation pathway composed of electron rich components led to effective charge migration from donor to acceptor in this molecule.7 Also the significant contribution from the π–π* transition at the longer wavelength region also worked beneficial for the dye.42 The electron lifetime extracted from the Bode phase plots (Fig. 13(b)) is consistent with the observed trend in open circuit voltage (VOC).
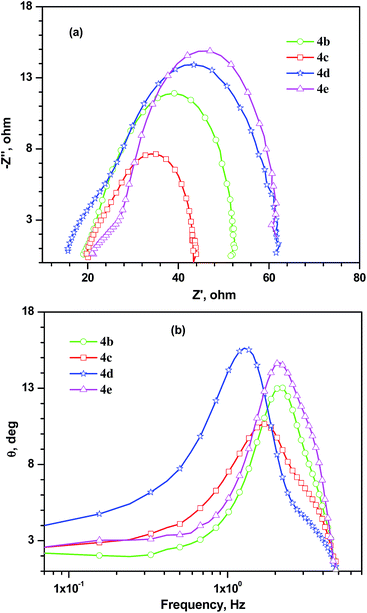 |
| | Fig. 13 Nyquist (a) and Bode-phase (b) plots observed for the DSSCs measured under illumination. | |
Conclusions
In conclusion, we have designed and synthesized a series of dyes containing diphenylaminocarbazole donor, cyanoacrylic acid acceptor and conjugation pathway containing different aryl/heteroaryl units such as phenyl, thiophene, fluorene and carbazole. Fluorene and carbazole containing dyes exhibited intense absorptions with high molar extinction coefficients while the terthiophene linked dye showed red-shifted charge transfer transition. TDDFT calculations showed the contribution of π-type orbital for the longer wavelength absorption. Interestingly the dye possessing more contribution from the π-type orbital to the visible region absorption led to efficient photocurrent generation. The thiophene, fluorene and carbazole containing dyes displayed low oxidation potentials than the phenyl analogues reflecting the electron richness of the conjugation pathway. Overall the dye containing terthiophene linker, 4c exhibited higher power conversion efficiency (4.88%) in the series attributable to its broader absorption in the visible region and favorable LUMO energy which led to efficient charge injection. Though the dyes possessing different linkers showed similar ground state oxidation potentials, they displayed huge variations in the excited state redox potentials which reflected in the charge collection efficiency of the devices and overall performance. Our studies point that the variation in the conjugation pathway is an effective method to modulate the excited state electronic properties and fine-tune the interfacial charge transport characteristics.
Experimental
General experimental methods
All the chemicals were purchased from readily available commercial sources and used as such without further purification. All the solvents were dried by standard procedures prior to use. Column chromatography purifications were performed with the use of silica gel (230–400 mesh) as a stationary phase in a column with 40 cm long and 3.0 cm diameter. The IR spectra were recorded with a NEXUS FT-IR spectrometer (Thermo nicolet) by using KBr pellets. The 1H and 13C NMR spectra were recorded with a Bruker (Bruker Avance 3) spectrometer operating at 500.13 and 125.77 MHz respectively. Deuterated chloroform (CDCl3) and dimethyl sulfoxide (DMSO-d6) were used as solvent with residual peak at δ 7.26 and 2.52 for 1H; 77.0 and 39.5 for 13C, respectively. UV-Vis spectra were recorded in Tol, DCM, CH, DMF, ACN, THF solvents at room temperature in quartz cuvettes using a Cary spectrophotometer (Cary 100). Emission spectra were recorded using a spectro fluorophotometer at room temperature. The cyclic voltammetry (CV) and differential pulse voltammetry (DPV) performed with epsilon (Basi epsilon) electrochemical analyzer instrument using a glassy carbon working electrode, a non-aqueous Ag/AgNO3 reference electrode. The experiments were performed at room temperature under nitrogen atmosphere in dichloromethane, Bu4NHClO4 as supporting electrolyte (0.1 M). The high resolution mass spectra were obtained from a Bruker Daltoniks GmBH (micrOTOF-QII) ESI mass spectrometer in the positive ion mode.
Synthesis
Synthesis of 7-bromo-9-butyl-N,N-diphenyl-9H-carbazol-2-amine8f (1), 5′-bromo-2,2′-bithiophene-5-carbaldehyde14a and 7-bromo-9,9-diethyl-9H-fluorene-2-carbaldehyde14b were prepared to according to the reported procedure.
9-Butyl-N,N-diphenyl-7-(thiophen-2-yl)-9H-carbazol-2-amine, 2. A mixture of 7-bromo-9-butyl-N,N-diphenyl-9H-carbazol-2-amine (1, 1.05 g, 4 mmol), tributyl(thiophen-2-yl)stannane (1.87 g, 4.8 mmol), Pd(PPh3)2Cl2 (0.03 g, 0.04 mmol) and DMF (5 mL) were sequentially charged into a two neck flask under N2 atmosphere and heated at 80 °C for 15 h. After cooling the reaction mixture was poured into water. The organic compound extracted with dichloromethane (3 × 40 mL). The combined organic extracts washed with brine solution, dried over Na2SO4. Further the crude product was purified by column chromatography on silica gel by using hexanes–dichloromethane mixture (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as an eluent. White solid; yield 1.71 g (91%); mp 119 °C; 1H NMR (CDCl3, 500.13 MHz) δ 7.96 (d, J = 7.5 Hz, 1H), 7.91 (d, J = 8.5 Hz, 1H), 7.54 (d, J = 1.0 Hz, 1H), 7.47 (dd, J = 8.5 Hz, 1.5 Hz, 1H), 7.40 (dd, J = 3.5 Hz, 1.0 Hz, 1H), 7.24–7.30 (m, 6H), 7.15–7.17 (m, 4H), 7.01–7.13 (m, 2H), 7.01 (dt, J = 7.5 Hz, 1.5 Hz, 2H), 6.97 (dd, J = 15 Hz, 1.5 Hz, 1H), 4.17 (t, J = 7.0 Hz, 2H), 1.75–1.78 (m, 2H), 1.29–1.34 (m, 2H), 0.89 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 125.77 MHz) δ 148.3, 146.4, 145.8, 142.2, 141.3, 131.3, 129.2, 128.0, 124.4, 123.9, 122.8, 122.5, 120.9, 120.1, 118.5, 117.8, 117.3, 105.9, 105.0, 42.6, 31.1, 20.5, 13.9; HRMS calcd for C32H28N2S [M]+ m/z 472.1973, found 472.1976.
:1) as an eluent. White solid; yield 1.71 g (91%); mp 119 °C; 1H NMR (CDCl3, 500.13 MHz) δ 7.96 (d, J = 7.5 Hz, 1H), 7.91 (d, J = 8.5 Hz, 1H), 7.54 (d, J = 1.0 Hz, 1H), 7.47 (dd, J = 8.5 Hz, 1.5 Hz, 1H), 7.40 (dd, J = 3.5 Hz, 1.0 Hz, 1H), 7.24–7.30 (m, 6H), 7.15–7.17 (m, 4H), 7.01–7.13 (m, 2H), 7.01 (dt, J = 7.5 Hz, 1.5 Hz, 2H), 6.97 (dd, J = 15 Hz, 1.5 Hz, 1H), 4.17 (t, J = 7.0 Hz, 2H), 1.75–1.78 (m, 2H), 1.29–1.34 (m, 2H), 0.89 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 125.77 MHz) δ 148.3, 146.4, 145.8, 142.2, 141.3, 131.3, 129.2, 128.0, 124.4, 123.9, 122.8, 122.5, 120.9, 120.1, 118.5, 117.8, 117.3, 105.9, 105.0, 42.6, 31.1, 20.5, 13.9; HRMS calcd for C32H28N2S [M]+ m/z 472.1973, found 472.1976.
3-(5-(9-Butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)benzaldehyde, 3a. A solution of 9-butyl-N,N-diphenyl-7-(thiophen-2-yl)-9H-carbazol-2-amine (2, 4.72 g, 10 mmol) in dry tetrahydrofuran (60 mL) was cooled to −78 °C. A solution of n-butyl lithium (7.0 mL, 1.6 M) added to it with vigorous stirring over 20 minutes. After the addition was over, the resulting mixture was stirred for another 2 h at this temperature and Bu3SnCl (3.91 g, 12 mmol) was added and the solution stirred at room temperature overnight. The reaction was quenched by the addition of cold water and the organic product extracted with chloroform (3 × 30 mL). The combined organic extract dried over anhydrous K2CO3, and concentrated to leave compound 3 in a quantitative yield. A mixture of 9-butyl-N,N-diphenyl-7-(5-(tributylstannyl)thiophen-2-yl)-9H-carbazol-2-amine (1.05 g, 1.2 mmol), 3-bromobenzaldehyde (0.19 g, 1 mmol) was dissolved in dry DMF (5 mL) and degassed with nitrogen, and added Pd(PPh3)2Cl2 (0.01 g, 0.01 mmol). The reaction mixture was heated at 80 °C for 15 h. After cooling the reaction mixture was poured into water. The organic compound extracted with dichloromethane (3 × 40 mL) and dried over Na2SO4. Further the crude product was purified by column chromatography on silica gel by using hexanes–dichloromethane mixture (2:1) as an eluent. Yellow solid; yield 0.55 g (96%); mp 151 °C; IR (KBr, cm−1) 1706 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (CDCl3, 500.13 MHz) δ 10.08 (s, 1H), 8.16 (t, J = 1.5 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.92 (dd, J = 8.0 Hz, 2.5 Hz, 2H), 7.78–7.80 (m, 1H), 7.56–7.59 (m, 2H), 7.51 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 7.42 (dd, J = 11.0 Hz, 3.5 Hz, 2H), 7.27–7.28 (m, 2H), 7.25 (s, 1H), 7.16–7.17 (m, 4H), 7.10 (d, J = 2.0 Hz, 1H), 7.01–7.04 (m, 3H), 6.98 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 4.19 (t, J = 7.0 Hz, 2H), 1.77–1.80 (m, 2H), 1.31–1.35 (m, 2H), 0.90 (m, 3H); 13C NMR (CDCl3, 125.77 MHz) δ 192.3, 148.4, 146.7, 146.4, 142.5, 141.4, 141.3, 137.2, 135.7, 131.3, 130.9, 129.8, 129.4, 128.8, 126.3, 125.2, 124.2, 124.1, 123.0, 122.8, 121.1, 120.3, 118.6, 117.6, 117.5, 105.8, 105.1, 42.9, 31.3, 20.7, 14.1; HRMS calcd for C39H32N2OS [M]+ m/z 576.2229, found 576.2232.
O); 1H NMR (CDCl3, 500.13 MHz) δ 10.08 (s, 1H), 8.16 (t, J = 1.5 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.92 (dd, J = 8.0 Hz, 2.5 Hz, 2H), 7.78–7.80 (m, 1H), 7.56–7.59 (m, 2H), 7.51 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 7.42 (dd, J = 11.0 Hz, 3.5 Hz, 2H), 7.27–7.28 (m, 2H), 7.25 (s, 1H), 7.16–7.17 (m, 4H), 7.10 (d, J = 2.0 Hz, 1H), 7.01–7.04 (m, 3H), 6.98 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 4.19 (t, J = 7.0 Hz, 2H), 1.77–1.80 (m, 2H), 1.31–1.35 (m, 2H), 0.90 (m, 3H); 13C NMR (CDCl3, 125.77 MHz) δ 192.3, 148.4, 146.7, 146.4, 142.5, 141.4, 141.3, 137.2, 135.7, 131.3, 130.9, 129.8, 129.4, 128.8, 126.3, 125.2, 124.2, 124.1, 123.0, 122.8, 121.1, 120.3, 118.6, 117.6, 117.5, 105.8, 105.1, 42.9, 31.3, 20.7, 14.1; HRMS calcd for C39H32N2OS [M]+ m/z 576.2229, found 576.2232.
4-(5-(9-Butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)benzaldehyde, 3b. Compound 3b was synthesized from 9-butyl-N,N-diphenyl-7-(5-(tributylstannyl)thiophen-2-yl)-9H-carbazol-2-amine (0.45 g, 0.6 mmol) and 4-bromobenzaldehyde (0.09 g, 0.5 mmol) by following a procedure similar to that described above for 3a. Yellow solid; yield 0.27 g (94%); mp 155 °C; IR (KBr, cm−1) 1695 (νCO); 1H NMR (CDCl3, 500.13 MHz) δ 10.00 (s, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.89–7.92 (m, 3H), 7.79 (d, J = 11.0 Hz, 2H), 7.56 (s, 1H), 7.51–7.47 (m, 2H), 7.41 (d, J = 3.5 Hz, 1H), 7.25–7.28 (m, 3H), 7.16 (d, J = 8.0 Hz, 4H), 7.10 (s, 1H), 7.02 (t, J = 7.0 Hz, 2H), 6.98 (d, J = 8.5 Hz, 1H), 4.17 (t, J = 7.5 Hz, 2H), 1.76–1.80 (m, 2H), 1.30–1.34 (m, 2H), 0.89 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 125.77 MHz) δ 191.4, 148.2, 147.5, 146.7, 142.3, 141.2, 141.1, 140.2, 134.9, 130.5, 129.2, 126.2, 125.6, 124.12, 124.06, 123.0, 122.6, 121.0, 120.2, 118.3, 117.4, 117.3, 105.6, 104.8, 42.7, 31.1, 20.5, 13.9; HRMS calcd for C39H32N2OSNa [M + Na]+ m/z 599.2127, found 599.2125.
5-(5-(5-(9-Butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)thiophen-2-yl)thiophene-2-carbaldehyde, 3c. Compound 3c was synthesized from 9-butyl-N,N-diphenyl-7-(5-(tributylstannyl)thiophen-2-yl)-9H-carbazol-2-amine (0.45 g, 0.6 mmol) and 5′-bromo-2,2′-bithiophene-5-carbaldehyde (0.14 g, 0.5 mmol) by following a procedure similar to that described above for 3a. Red solid; yield 0.31 g (94%); mp 182 °C; IR (KBr, cm−1) 1687 (νCO); 1H NMR (CDCl3, 500.13 MHz) δ 9.87 (s, 1H), 7.97 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.68 (d, J = 4.0 Hz, 1H), 7.52 (s, 1H), 7.47 (d, J = 8.0 Hz, 1H), 7.33–7.34 (m, 2H), 7.26–7.28 (m, 4H), 7.24–7.25 (m, 2H), 7.15–7.18 (m, 5H), 7.10 (s, 1H), 7.02 (t, J = 7.5 Hz, 2H), 6.97 (dd, J = 8.5 Hz, 2.0 Hz, 1H), 4.18 (t, J = 6.5 Hz, 2H), 1.74–1.80 (m, 2H), 1.28–1.34 (m, 2H), 0.89 (t, J = 2.0 Hz, 3H); 13C NMR (CDCl3, 125.77 MHz) δ 182.5, 148.2, 146.9, 146.6, 145.8, 142.3, 141.5, 141.2, 139.5, 137.5, 135.1, 134.2, 130.5, 129.2, 127.1, 125.5, 124.32, 124.04, 123.99, 123.7, 122.9, 122.6, 120.9, 120.2, 118.3, 117.4, 117.3, 105.5, 104.8, 42.7, 31.1, 20.5, 13.9; HRMS calcd for C41H32N2OS3 [M]+ m/z 664.1676, found 664.1654.
7-(5-(9-Butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)-9,9-diethyl-9H-fluorene-2-carbaldehyde, 3d. Compound 3d was synthesized from 9-butyl-N,N-diphenyl-7-(5-(tributylstannyl)thiophen-2-yl)-9H-carbazol-2-amine (0.45 g, 0.6 mmol) and 7-bromo-9,9-diethyl-9H-fluorene-2-carbaldehyde (0.16 g, 0.5 mmol) by following a procedure similar to that described above for 3a. Orange solid; yield 0.35 g (97%); mp 160 °C; IR (KBr, cm−1) 1691 (νCO); 1H NMR (CDCl3, 500.13 MHz) δ 10.06 (s, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.87 (dd, J = 6.0 Hz, 1.5 Hz, 2H), 7.84 (d, J = 8.0 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.71 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 7.64 (d, J = 1.5 Hz, 1H), 7.58 (d, J = 1.0 Hz, 1H), 7.52 (dd, J = 8.5 Hz, 1.5 Hz, 1H), 7.43 (d, J = 3.5 Hz, 1H), 7.42 (d, J = 7.0 Hz, 1H), 7.27–7.29 (m, 3H), 7.25 (s, 1H), 7.16 (dd, J = 3.5 Hz, 1.0 Hz, 4H), 7.10 (d, J = 1.5 Hz, 1H), 7.01–7.04 (m, 2H), 6.98 (dd, J = 7.5 Hz, 2.0 Hz, 1H), 4.19 (t, J = 7.0 Hz, 2H), 2.12–2.17 (m, 4H), 1.78–1.81 (m, 2H), 1.28–1.36 (m, 2H), 0.87–0.92 (m, 3H), 0.36 (t, J = 7.5 Hz, 6H); 13C NMR (CDCl3, 125.77 MHz) δ 192.5, 152.5, 151.0, 148.4, 147.7, 146.7, 145.7, 143.3, 142.4, 141.4, 139.5, 135.4, 135.2, 131.2, 130.9, 129.4, 125.0, 124.7, 124.2, 124.1, 123.3, 122.9, 122.7, 121.6, 121.1, 120.3, 120.10, 120.06, 118.6, 117.6, 117.4, 105.7, 105.1, 56.6, 42.9, 32.9, 31.3, 20.7, 14.1, 8.7; HRMS calcd for C50H44N2OS [M]+ m/z 720.3174, found 720.3163.
9-Butyl-7-(5-(9-butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)-9H-carbazole-2-carbaldehyde, 3e. Compound 3d was synthesized from 9-butyl-N,N-diphenyl-7-(5-(tributylstannyl)thiophen-2-yl)-9H-carbazol-2-amine (0.45 g, 0.6 mmol) and 7-bromo-9-butyl-9H-carbazole-2-carbaldehyde 0.17 g (0.5 mmol) by following a procedure similar to that described above for 3a. Yellow solid; yield (0.34 g, 93%); mp 172 °C; IR (KBr, cm−1) 1691 (νCO); 1H NMR (CDCl3, 500.13 MHz) δ 10.16 (s, 1H), 8.13–8.19 (m, 2H), 7.91–8.00 (m, 3H), 7.75 (d, J = 7.5 Hz, 1H), 7.67 (s, 1H), 7.53–7.62 (m, 3H), 7.43–7.47 (m, 3H), 7.28 (s, 2H), 7.16 (d, J = 6.0 Hz, 4H), 7.11 (s, 2H), 6.98–7.04 (m, 3H), 4.41 (m, 2H), 4.19 (m, 2H), 1.92–1.93 (m, 2H), 1.78–1.79 (m, 2H), 1.44–1.47 (m, 2H), 1.32–1.34 (m, 2H), 0.98–1.00 (m, 3H), 0.89–0.91 (m, 3H); 13C NMR (CDCl3, 125.77 MHz) δ 192.6, 148.2, 146.5, 145.6, 143.7, 142.8, 142.3, 141.3, 140.8, 134.1, 133.8, 131.6, 129.2, 128.0, 124.7, 124.0, 123.9, 122.7, 122.6, 121.9, 121.7, 121.3, 120.9, 120.5, 120.2, 118.5, 118.0, 117.4, 117.3, 109.6, 105.8, 105.5, 104.9, 43.2, 42.7, 31.3, 31.1, 20.6, 20.5, 13.9; HRMS calcd for C49H43N3OS [M]+ m/z 721.3126, found 721.3121.
(E)-3-(3-(5-(9-Butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)phenyl)-2-cyanoacrylic acid, 4a. A mixture of 3-(5-(9-butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)benzaldehyde (3a, 0.19 g, 0.33 mmol), 2-cyanoacetic acid (0.04 g, 0.5 mmol), ammonium acetate (0.04 g, 0.25 mmol), and acetic acid (5 mL) refluxed for 24 h. The resulting yellow precipitate was filtered, washed several times with water and dried under vacuum. It was crystallized from dichloromethane–hexanes mixture to obtain the analytically pure sample. Brown solid; yield 0.19 g (89%); mp 125 °C; IR (KBr, cm−1) 2221 (νC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N); 1H NMR (DMSO-d6, 500.13 MHz) δ 8.30 (s, 1H), 8.05–8.11 (m, 2H), 7.88 (s, 2H), 7.59–7.72 (m, 3H), 7.49–7.53 (m, 2H), 7.31–7.34 (m, 4H), 7.20 (s, 1H), 7.04–7.09 (m, 7H), 6.88 (d, J = 8.0 Hz, 1H), 4.30–4.32 (m, 2H), 1.68 (m, 2H), 1.21–1.23 (m, 2H), 0.82 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 125.77 MHz) δ 147.6, 145.8, 144.6, 141.8, 140.9, 130.4, 129.4, 126.6, 124.7, 124.2, 123.5, 122.7, 121.2, 120.3, 118.6, 117.9, 116.9, 116.7, 105.4, 104.9, 41.70, 30.6, 19.7, 13.6; HRMS calcd for C42H33N3O2S [M]+ m/z 666.2186, found 666.2187.
N); 1H NMR (DMSO-d6, 500.13 MHz) δ 8.30 (s, 1H), 8.05–8.11 (m, 2H), 7.88 (s, 2H), 7.59–7.72 (m, 3H), 7.49–7.53 (m, 2H), 7.31–7.34 (m, 4H), 7.20 (s, 1H), 7.04–7.09 (m, 7H), 6.88 (d, J = 8.0 Hz, 1H), 4.30–4.32 (m, 2H), 1.68 (m, 2H), 1.21–1.23 (m, 2H), 0.82 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 125.77 MHz) δ 147.6, 145.8, 144.6, 141.8, 140.9, 130.4, 129.4, 126.6, 124.7, 124.2, 123.5, 122.7, 121.2, 120.3, 118.6, 117.9, 116.9, 116.7, 105.4, 104.9, 41.70, 30.6, 19.7, 13.6; HRMS calcd for C42H33N3O2S [M]+ m/z 666.2186, found 666.2187.
(E)-3-(4-(5-(9-Butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)phenyl)-2-cyanoacrylic acid, 4b. It was prepared from 4-(5-(9-butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)benzaldehyde (3b, 0.19 g, 0.33 mmol) and 2-cyano acetic acid (0.04 g, 0.5 mmol) by following a procedure described above for 4a. Red solid; yield 0.17 g (82%); mp 205 °C; IR (KBr, cm−1) 2216 (νCN); 1H NMR (DMSO-d6, 500.13 MHz) δ 8.20 (s, 1H), 8.11 (d, J = 8.0 Hz, 1H), 8.07 (d, J = 8.0 Hz, 3H), 7.92 (t, J = 7.0 Hz, 3H), 7.80 (d, J = 3.5 Hz, 1H), 7.75 (d, J = 4.0 Hz, 1H), 7.55 (d, J = 8.5 Hz, 1H), 7.33 (t, J = 7.5 Hz, 4H), 7.18 (d, J = 1.5 Hz, 1H), 7.05–7.10 (m, 6H), 6.89 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 4.31 (t, J = 6.5 Hz, 2H), 1.66–1.71 (m, 2H), 1.21–1.25 (m, 2H), 0.83 (t, J = 7.0 Hz, 3H); 13C NMR (DMSO-d6, 125.77 MHz) δ 148.1, 146.6, 146.4, 142.4, 141.4, 141.0, 138.1, 132.0, 130.9, 130.6, 129.9, 127.7, 125.8, 125.7, 124.0, 123.2, 118.3, 117.5, 117.2, 106.1, 105.4, 42.2, 31.1, 20.2, 14.1; HRMS calcd for C42H33N3O2S [M]+ m/z 643.2287, found 643.2288.
(E)-3-(5-(5-(5-(9-Butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)thiophen-2-yl)thiophen-2-yl)-2-cyanoacrylic acid, 4c. It was prepared from 5-(5-(5-(9-Butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)thiophen-2-yl)thiophene-2-carbaldehyde (3c, 0.33 g, 0.5 mmol) and 2-cyanoacetic acid (0.06 g, 0.7 mmol) by following a procedure described above for 4a. Black solid. Yield 0.32 g (87%); mp 212 °C; IR (KBr, cm−1) 2216 (νCN); 1H NMR (DMSO-d6, 500.13 MHz) δ 8.50 (s, 1H), 8.05–8.10 (m, 2H), 8.00 (d, J = 4.0 Hz, 2H), 7.87 (s, 1H), 7.69 (d, J = 4.0 Hz, 1H), 7.63 (dd, J = 6.5 Hz, 4.0 Hz, 2H), 7.50–7.54 (m, 2H), 7.46 (d, J = 4.0 Hz, 1H), 7.33 (t, J = 8.5 Hz, 4H), 7.17 (d, J = 1.5 Hz, 1H), 7.05–7.09 (m, 6H), 4.30 (t, J = 7.0 Hz, 2H), 1.64–1.70 (m, 2H), 1.18–1.25 (m, 2H), 0.78 (t, J = 7.5 Hz, 3H); 13C NMR (DMSO-d6, 125.77 MHz) δ 163.6, 147.6, 146.1, 146.0, 145.1, 144.8, 141.9, 141.4, 140.9, 138.5, 134.2, 134.0, 133.4, 129.9, 129.4, 128.2, 126.4, 125.3, 125.0, 124.8, 123.6, 122.7, 122.1, 121.2, 120.4, 117.9, 117.0, 116.7, 116.6, 105.6, 104.9, 41.7, 30.6, 19.71, 13.65; HRMS calcd for C44H33N3O2S3, [M]+ m/z 731.1729, found 731.1729.
(E)-3-(7-(5-(9-Butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)-9,9-diethyl-9H-fluoren-2-yl)-2-cyanoacrylic acid, 4d. It was prepared from 7-(5-(9-butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)-9,9-diethyl-9H-fluorene-2-carbaldehyde (3d, 0.36 g, 0.5 mmol) and 2-cyanoacetic acid (0.06 g, 0.7 mmol) by following a procedure described above for 4a. Red solid; yield 0.37 g (93%); mp 138 °C; IR (KBr, cm−1) 2216 (νCN); 1H NMR (DMSO-d6, 500.13 MHz) δ 8.37 (s, 1H), 8.16 (s, 1H), 8.05–8.11 (m, 4H), 8.00 (d, J = 8.0 Hz, 1H), 7.89 (d, J = 10.0 Hz, 2H), 7.79–7.78 (m, 1H), 7.72–7.76 (m, 2H), 7.55–7.57 (m, 1H), 7.31–7.34 (t, J = 8.0 Hz, 4H), 7.17 (d, J = 1.5 Hz, 1H), 7.05–7.10 (m, 6H), 6.87–6.89 (m, 1H), 4.31 (t, J = 6.0 Hz, 2H), 2.08–2.19 (m, 4H), 1.67–1.70 (m, 2H), 1.20–1.26 (m, 2H), 0.81–0.84 (m, 3H), 0.30–0.34 (m, 6H); 13C NMR (DMSO-d6, 125.77 MHz) δ 152.2, 150.7, 148.1, 146.3, 144.7, 142.7, 142.3, 141.4, 139.6, 134.6, 131.0, 129.9, 126.0, 125.4, 125.2, 125.0, 124.0, 123.2, 122.4, 122.2, 121.7, 121.0, 120.8, 119.9, 118.4, 117.4, 117.2, 105.9, 105.5, 56.4, 42.2, 32.1, 31.1, 20.2, 14.1, 8.9; HRMS calcd for C53H45N3O2S [M]+ m/z 787.3227, found 787.3224.
(E)-3-(9-Butyl-7-(5-(9-butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)-9H-carbazol-2-yl)-2-cyanoacrylic acid, 4e. It was prepared from 9-butyl-7-(5-(9-butyl-7-(diphenylamino)-9H-carbazol-2-yl)thiophen-2-yl)-9H-carbazole-2-carbaldehyde (3e, 0.24 g, 0.5 mmol) and 2-cyanoacetic acid (0.04 g, 0.5 mmol) by following a procedure described above for 4a. Red solid; yield 0.34 g (86%); mp 234 °C; IR (KBr, cm−1) 2217 (νCN); 1H NMR (DMSO-d6, 500.13 MHz) δ 8.52 (s, 1H), 8.36 (s, 1H), 8.25–8.32 (m, 2H), 8.02–8.07 (m, 3H), 8.00 (d, J = 8.0 Hz, 1H), 7.89 (s, 1H), 7.73–7.79 (m, 2H), 7.62 (d, J = 8.0 Hz, 1H), 7.53 (d, J = 7.5 Hz, 1H), 7.30–7.32 (m, 4H), 7.17 (s, 1H), 7.03–7.08 (m, 6H), 6.86 (d, J = 7.5 Hz, 1H), 4.48–4.49 (m, 2H), 4.28–4.30 (m, 2H), 1.82–1.84 (m, 2H), 1.65–1.67 (m, 2H), 1.34–1.37 (m, 2H), 1.20–1.23 (m, 2H), 0.92 (t, J = 7.5 Hz, 3H), 0.81–0.82 (m, 3H); 13C NMR (DMSO-d6, 125.77 MHz) δ 163.7, 155.1, 147.6, 145.8, 144.3, 142.8, 142.3, 141.8, 140.9, 140.2, 133.2, 130.5, 129.4, 128.5, 125.7, 125.6, 124.8, 123.5, 122.7, 122.0, 121.8, 121.2, 121.1, 120.8, 120.7, 120.3, 117.9, 117.5, 116.9, 116.6, 112.8, 105.6, 105.3, 104.9, 42.3, 41.7, 30.7, 30.6, 19.8, 19.7, 13.7, 13.6; HRMS calcd for C52H44N4O2S [M]+ m/z 788.3179, found 788.3151.
Computational methods
Gaussian 09 program package was used to perform all the computations.43 The ground-state geometries were fully optimized without any symmetry constraints using DFT employing a Becke's hybrid correlation functional B3LYP31 with 6-31G (d,p) basis set for all atoms. Vibrational analysis on the optimized structures was performed to confirm the structure. The excitation energies and oscillator strengths for the lowest 10 singlet–singlet transitions at the optimized geometry in the ground state were obtained by TD-DFT calculations using the same basis set with two different kinds of hybrid functionals namely BMK33/dgdzvp34 and MPW1K/6-31G(d,p).32 For the calculations using BMK33 functional and dgdzvp34 basis set the SMD36 solvation model was adopted. But for the calculations using MPW1K32 functional PCM35 solvation was applied.
Device fabrication and characterization
A fluorine-doped SnO2 conducting glass (FTO, 7 Ω sq−1, transmittance ∼80%, NSG America, Inc., New Jersey, USA) was first cleaned with a neutral cleaner, and then washed with deionized water, acetone, and isopropyl alcohol, sequentially. The conducting surface of the FTO was treated with a solution of titanium tetraisopropoxide (1 g) in 2-methoxyethanol (3 g) for obtaining a good mechanical contact between the conducting glass and TiO2 film, as well as to isolate the conducting glass surface from the electrolyte. TiO2 paste was coated onto the treated conducting glass by rolling a metal strip over it (doctor blade technique). After coating each TiO2 layer, the dried TiO2 film was gradually heated to 450 °C in an oxygen atmosphere, and subsequently sintered at that temperature for 30 min. The TiO2 photo-anodes of the DSSCs employed in the experiments were composed of a 14 μm thick transparent TiO2 layer with a scattering layer of 4.5 μm thickness. After sintering at 450 °C and cooling to 80 °C, the TiO2 film was immersed in a 3 × 10−4 M solution of dye at room temperature for 24 h. N7192 (Solaronix S.A., Aubonne, Switzerland) was dissolved in acetonitrile (ACN) and tert-butyl alcohol (volume ratio of 1:1) and used as a standard dye solution for device optimization. The solutions of the dyes were prepared in a mixing solvent containing ACN, tert-butyl alcohol and dimethyl sulfoxide (DMSO) (volume ratio of 1:1:3). Then the photoanode (FTO/TiO2/dye) was placed on a platinum-sputtered conducting glass electrode (ITO, 7 Ω sq−1, Ritek Corporation, Hsinchu, Taiwan), keeping the two electrodes separated by a 25 μm thick surlyn® (SX1170-25, Solaronix S.A., Aubonne, Switzerland). The two electrodes were then sealed by heating. A mixture of 0.1 M LiI, 0.6 M 1-propyl-2,3-dimethylimidazolium iodide (DMPII), 0.05 M I2, and 0.5 M tert-butylpyridine (TBP) in 3-methoxypropionitrile (MPN)/CAN (volume ratio of 1:1) was used as the electrolyte. The electrolyte was injected into the gap between the electrodes by capillarity; the electrolyte-injecting hole was previously made in the counter electrode with a drilling machine, and the hole was sealed with hot-melt glue after the injection of the electrolyte. Surface of the DSSC was covered by a mask with a light illuminated area of 0.16 cm2 and then illuminated by a class-A quality solar simulator (XES-301S, AM 1.5 G, San-Ei Electric Co., Ltd.). Incident light intensity (100 mW cm−2) was calibrated with a standard Si Cell (PECSI01, Peccell Technologies, Inc.). Photocurrent–voltage curves of the DSSCs were obtained with a potentiostat/galvanostat (PGSTAT 30, Autolab, Eco-Chemie, The Netherlands). The thickness of the TiO2 film was judged by scanning electron microscopic images (SEM, NanoSEM 230, Novat). For UV-absorption spectra, dye molecules were coated on the TiO2 films and the corresponding spectra were obtained using an UV-visible spectrophotometer (V-570, Jasco, Japan) equipped with a total integrating sphere. Electrochemical impedance spectra (EIS) were obtained by the above-mentioned potentiostat/galvanostat, equipped with an FRA2 module, under a constant light illumination of 100 mW cm−2. The frequency range explored was 10 mHz to 65 kHz. The applied bias voltage was set at the open circuit voltage of the DSSC, between the ITO-Pt counter electrode and the FTO–TiO2-dye working electrode, starting from the short circuit conditions; the corresponding AC amplitude was 10 mV. The impedance spectra were analyzed using an equivalent circuit model. Incident photo-to-current conversion efficiency (IPCE) curves were obtained under short-circuit conditions. The light source was a class A quality solar simulator (PEC-L11, AM 1.5 G, Peccell Technologies, Inc.); light was focused through a monochromator (Oriel Instrument, model 74100) onto the photo-voltaic cell. The monochromator was incremented through the visible spectrum to generate the IPCE (λ) as defined by IPCE (λ) = 1240 (JSC/λφ), where λ is the wavelength, JSC is short-circuit photocurrent density (mA cm−2) recorded with a potentiostat/galvanostat, and is the incident radiative flux (W m−2) measured with an optical detector (Oriel Instrument, model 71580) and a power meter (Oriel Instrument, model 70310).
Acknowledgements
KRJT is thankful to DST, New Delhi for generous financial support (Ref. No. DST/TSG/PT/2013/09). AV acknowledges a research fellowship from UGC, New Delhi. We are also thankful to DST for the purchase of ESI mass spectrometer via the FIST grant to the Chemistry Department, IIT Roorkee.
Notes and references
-
(a) A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed
 ;
(b) Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 2903–2934 CrossRef CAS ;
(c) A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed ;
(d) Y. Ooyama and Y. Harima, ChemPhysChem, 2012, 13, 4032–4080 CrossRef CAS PubMed ;
(e) Y.-Z. Wu and W.-H. Zhu, Chem. Soc. Rev., 2013, 42, 2039–2058 RSC ;
(f) B.-G. Kim, K. Chung and J. Kim, Chem.–Eur. J., 2013, 19, 5220–5230 CrossRef CAS PubMed .
;
(b) Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 2903–2934 CrossRef CAS ;
(c) A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed ;
(d) Y. Ooyama and Y. Harima, ChemPhysChem, 2012, 13, 4032–4080 CrossRef CAS PubMed ;
(e) Y.-Z. Wu and W.-H. Zhu, Chem. Soc. Rev., 2013, 42, 2039–2058 RSC ;
(f) B.-G. Kim, K. Chung and J. Kim, Chem.–Eur. J., 2013, 19, 5220–5230 CrossRef CAS PubMed . -
(a) M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed ;
(b) M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Müller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS ;
(c) I. Stengel, N. Pootrakulchote, R. R. Dykeman, A. Mishra, S. M. Zakeeruddin, P. J. Dyson, M. Grätzel and P. Bäuerle, Adv. Energy Mater., 2012, 2, 1004–1012 CrossRef CAS ;
(d) C.-Y. Chen, N. Pootrakulchote, T.-H. Hung, C.-J. Tan, H.-H. Tsai, S. M. Zakeeruddin, C.-G. Wu and M. Grätzel, J. Phys. Chem. C, 2011, 115, 20043–20050 CrossRef CAS ;
(e) S. M. Feldt, E. A. Gibson, E. Gabrielsson, L. Sun, G. Boschloo and A. Hagfeldt, J. Am. Chem. Soc., 2010, 132, 16714–16724 CrossRef CAS PubMed ;
(f) A. Reynal and E. Palomares, Eur. J. Inorg. Chem., 2011, 4509–4526 CrossRef CAS .
-
(a) T. C. Sum and N. Mathews, Energy Environ. Sci., 2014, 7, 2518–3253 RSC ;
(b) J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 316–320 CrossRef CAS PubMed ;
(c) H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591 Search PubMed ;
(d) H. J. Snaith, J. Phys. Chem. Lett., 2013, 4, 3623–3630 CrossRef CAS ;
(e) M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed ;
(f) N. J. Jeon, J. Lee, J. H. Noh, M. K. Nazeeruddin, M. Grätzel and S. I. Seok, J. Am. Chem. Soc., 2013, 135, 19087–19090 CrossRef CAS PubMed .
-
(a) Z. Ning and H. Tian, Chem. Commun., 2009, 5483–5495 RSC ;
(b) M. Liangw and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC ;
(c) S. Cai, X. Hu, Z. Zhang, J. Su, X. Li, A. Islam, L. Han and H. Tian, J. Mater. Chem. A, 2013, 1, 4763–4772 RSC ;
(d) S. Cai, G. Tian, X. Li, J. Su and H. Tian, J. Mater. Chem. A, 2013, 1, 11295–11305 RSC ;
(e) D. P. Hagberg, T. Marinado, K. M. Karlsson, K. Nonomura, P. Qin, G. Boschloo, T. Brinck, A. Hagfeldt and L. Sun, J. Org. Chem., 2007, 72, 9550–9556 CrossRef CAS PubMed .
-
(a) A. Yella, R. Humphry-Baker, B. F. E. Curchod, N. A. Astani, J. Teuscher, L. E. Polander, S. Mathew, J.-E. Moser, I. Tavernelli, U. Rothlisberger, M. Grätzel, M. K. Nazeeruddin and J. Frey, Chem. Mater., 2013, 25, 2733–2739 CrossRef CAS ;
(b) L. E. Polander, A. Yella, J. Teuscher, R. Humphry-Baker, B. F. E. Curchod, N. A. Astani, P. Gao, J.-E. Moser, I. Tavernelli, U. Rothlisberger, M. Grätzel, M. K. Nazeeruddin and J. Frey, Chem. Mater., 2013, 25, 2642–2648 CrossRef CAS ;
(c) A. Venkateswararao, K. R. J. Thomas, C.-P. Lee and K.-C. Ho, Asian J. Org. Chem., 2015, 4, 69–80 CrossRef CAS ;
(d) M. Xu, M. Zhang, M. Pastore, R. Li, F. De Angelis and P. Wang, Chem. Sci., 2012, 3, 976–983 RSC .
-
(a) E. Gabrielsson, H. Ellis, S. Feldt, H. Tian, G. Boschloo, A. Hagfeldt and L. Sun, Adv. Energy Mater., 2013, 3, 1647–1656 CrossRef CAS ;
(b) D.-Y. Chen, Y.-Y. Hsu, H.-C. Hsu, B.-S. Chen, Y.-T. Lee, H. Fu, M.-W. Chung, S.-H. Liu, H.-C. Chen, Y. Chi and P.-T. Chou, Chem. Commun., 2010, 46, 5256–5258 RSC ;
(c) H. Qin, S. Wenger, M. Xu, F. Gao, X. Jing, P. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 130, 9202–9203 CrossRef CAS PubMed ;
(d) W. Zeng, Y. Cao, Y. Bai, Y. Wang, Y. Shi, M. Zhang, F. Wang, C. Pan and P. Wang, Chem. Mater., 2010, 22, 1915–1925 CrossRef CAS ;
(e) J. Zhang, Z. Yao, Y. Cai, L. Yang, M. Xu, R. Li, M. Zhang, X. Dong and P. Wang, Energy Environ. Sci., 2013, 6, 1604–1614 RSC .
-
(a) A. Baheti, P. Tyagi, K. R. J. Thomas, Y.-C. Hsu and J. T. Lin, J. Phys. Chem. Lett., 2009, 113, 8541–8547 CrossRef CAS ;
(b) Z.-S. Huang, H.-L. Feng, X.-F. Zang, Z. Iqbal, H. Zeng, D.-B. Kuang, L. Wang, H. Meier and D. Cao, J. Mater. Chem. A, 2014, 2, 15365–15376 RSC ;
(c) X. Hao, M. Liang, X. Cheng, X. Pian, Z. Sun and S. Xue, Org. Lett., 2011, 13, 5424–5427 CrossRef CAS PubMed ;
(d) P. Gao, H. N. Tsao, M. Grätzel and M. K. Nazeeruddin, Org. Lett., 2012, 14, 4330–4333 CrossRef CAS PubMed ;
(e) J.-H. Chen, C.-H. Tsai, S.-A. Wang, Y.-Y. Lin, T.-W. Huang, S.-F. Chiu, C.-C. Wu and K.-T. Wong, J. Org. Chem., 2011, 76, 8977–8985 CrossRef CAS PubMed ;
(f) N. Cai, R. Li, Y. Wang, M. Zhang and P. Wang, Energy Environ. Sci., 2013, 6, 139–147 RSC ;
(g) Y.-J. Cheng, C.-H. Chen, T.-Y. Lin and C.-S. Hsu, Chem.–Asian J., 2012, 7, 818–825 CrossRef CAS PubMed .
-
(a) K. R. J. Thomas and A. Baheti, Mater. Sci. Technol., 2013, 28, 71–87 Search PubMed ;
(b) S. Kim, J. K. Lee, S. O. Kang, J. Ko, J.-H. Yum, S. Fantacci, F. D. Angelis, D. D. Censo, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 16701–16707 CrossRef CAS PubMed ;
(c) A. Baheti, K. R. J. Thomas, C.-P. Lee and K.-C. Ho, Chem.–Asian J., 2012, 7, 2942–2954 CrossRef CAS PubMed ;
(d) A. Venkateswararao and K. R. J. Thomas, in Solar Cell Nanotechnology, ed. A. Tiwari, R. Boukherroub and M. Sharon, Wiley-Scrivener, Beverly, MA, 2014, ch. 2, pp. 41–96 Search PubMed ;
(e) A. Venkateswararao, K. R. J. Thomas, C.-P. Lee and K.-C. Ho, Tetrahedron Lett., 2013, 54, 3985–3989 CrossRef CAS ;
(f) A. Venkateswararao, K. R. J. Thomas, C.-P. Lee, C.-T. Li and K.-C. Ho, ACS Appl. Mater. Interfaces, 2014, 6, 2528–2539 CrossRef CAS PubMed .
-
(a) K. R. J. Thomas, J. T. Lin, Y.-T. Tao and C.-W. Ko, J. Am. Chem. Soc., 2001, 123, 9404–9411 CrossRef CAS PubMed ;
(b) T. Leijtens, I.-K. Ding, T. Giovenzana, J. T. Bloking, M. D. McGehee and A. Sellinger, ACS Nano, 2012, 6, 1455–1462 CrossRef CAS PubMed ;
(c) D. B. Romero, M. Schaer, M. Leclerc, D. Adès, A. Siove and L. Zuppiroli, Synth. Met., 1996, 80, 271–409 CrossRef CAS ;
(d) D. B. Romero, F. Nüesch, T. Benazzi, D. Adès, A. Siove and L. Zuppiroli, Adv. Mater., 1997, 9, 1158–1161 CrossRef CAS .
-
(a) P. Gao, H. N. Tsao, C. Yi, M. Grätzel and M. K. Nazeeruddin, Adv. Energy Mater., 2014, 4, 1301485 Search PubMed ;
(b) C. Teng, X. Yang, C. Yang, S. Li, M. Cheng, A. Hagfeldt and L. Sun, J. Phys. Chem. C, 2010, 114, 9101–9110 CrossRef CAS ;
(c) C.-C. Yu, K.-J. Jiang, J.-H. Huang, F. Zhang, X. Bao, F.-W. Wang, L.-M. Yang and Y. Song, Org. Electron., 2013, 14, 445–450 CrossRef CAS ;
(d) Q. Chai, W. Li, Y. Wu, K. Pei, J. Liu, Z. Geng, H. Tian and W. Zhu, ACS Appl. Mater. Interfaces, 2014, 6, 14621–14630 CrossRef CAS PubMed .
-
(a) N. Koumura, Z.-S. Wang, S. Mori, M. Miyashita, E. Suzuki and K. Hara, J. Am. Chem. Soc., 2006, 128, 14256–14257 CrossRef CAS PubMed ;
(b) H. Choi, J.-J. Kim, K. Song, J. Ko, M. K. Nazeeruddin and M. Grätzel, J. Mater.
Chem., 2010, 20, 3280–3286 CAS ;
(c) Y. Hao, X. C. Yang, M. Z. Zhou, J. Y. Cong, X. N. Wang, A. Hagfeldt and L. Sun, ChemSusChem, 2011, 4, 1601–1605 CrossRef CAS PubMed ;
(d) Z.-S. Wang, N. Koumura, Y. Cui, M. Takahashi, H. Sekiguchi, A. Mori, T. Kubo, A. Furube and K. Hara, Chem. Mater., 2008, 20, 3993–4003 CrossRef CAS ;
(e) X. Hu, S. Cai, G. Tian, X. Li, J. Su and J. Li, RSC Adv., 2013, 3, 22544–22553 RSC ;
(f) Y. Cui, Y. Wu, X. Lu, X. Zhang, G. Zhou, F. B. Miapeh, W. Zhu and Z.-S. Wang, Chem. Mater., 2011, 23, 4394–4401 CrossRef CAS ;
(g) S. Haid, M. Marszalek, A. Mishra, M. Wielopolski, T. Joél, J.-E. Moser, R. Humphry-Baker, S. M. Zakeeruddin, M. Grätzel and P. Bäuerle, Adv. Funct. Mater., 2012, 22, 1291–1302 CrossRef CAS ;
(h) T. Duan, K. Fan, C. Zhong, T. Peng, J. Qin and X. Chen, RSC Adv., 2012, 2, 7081–7086 RSC .
-
(a) T. Khanasa, N. Jantasing, S. Morada, N. Leesakul, R. Tarsang, S. Namuangruk, T. Kaewin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, Eur. J. Org. Chem., 2013, 2608–2620 CrossRef CAS ;
(b) P. Shen, Y. Liu, X. Huang, B. Zhao, N. Xiang, J. Fei, L. Liu, X. Wang, H. Huang and S. Tan, Dyes Pigm., 2009, 83, 187–197 CrossRef CAS ;
(c) K. R. J. Thomas, Y.-C. Hsu, J. T. Lin, K.-M. Lee, K.-C. Ho, C.-H. Lai, Y.-M. Cheng and P.-T. Chou, Chem. Mater., 2008, 20, 1830–1840 CrossRef CAS ;
(d) Y. J. Chang and T. J. Chow, Tetrahedron, 2009, 65, 4726–4734 CrossRef CAS ;
(e) A. Baheti, C.-P. Lee, K. R. J. Thomas and K.-C. Ho, Phys. Chem. Chem. Phys., 2011, 13, 17210–17221 RSC ;
(f) K. R. J. Thomas, J. T. Lin, Y.-C. Hsu and K.-C. Ho, Chem. Commun., 2005, 4098–4100 RSC ;
(g) H. Lai, J. Hong, P. Liu, C. Yuan, Y. Li and Q. Fang, RSC Adv., 2012, 2, 2427–2432 RSC .
- J. K. Stille, Angew. Chem., Int. Ed., 1986, 25, 508–524 CrossRef .
-
(a) H.-C. Chu, D. Sahu, Y.-C. Hsu, H. Padhy, D. Patra, J.-T. Lin, D. Bhattacharya, K.-L. Lu, K.-H. Wei and H.-C. Lin, Dyes Pigm., 2012, 93, 1488–1497 CrossRef CAS ;
(b) A. R. Morales, A. Frazer, A. W. Woodward, H.-Y. Ahn-White, A. Fonari, P. Tongwa, T. Timofeeva and K. D. Belfield, J. Org. Chem., 2013, 78, 1014–1025 CrossRef CAS PubMed .
- E. Knoevenagel, Chem. Ber., 1896, 29, 172–174 CrossRef CAS .
- K. Panthi, P. Z. El-Khoury, A. N. Tarnovsky and T. H. Kinstle, Tetrahedron, 2010, 66, 9641–9649 CrossRef CAS .
-
(a) Z. R. Grabowski and K. Rotkiewicz, Chem. Rev., 2003, 103, 3899–4031 CrossRef PubMed ;
(b) D.-F. Feng and L. Kevan, Chem. Rev., 1980, 80, 1–20 CrossRef CAS ;
(c) M. Glasbeek and H. Zhang, Chem. Rev., 2004, 104, 1929–1954 CrossRef CAS PubMed .
-
(a) O. van den Berg, W. F. Jager and S. J. Picken, J. Org. Chem., 2006, 71, 2666–2676 CrossRef CAS PubMed ;
(b) A. Granzhan, H. Ihmels and G. Viola, J. Am. Chem. Soc., 2007, 129, 1254–1267 CrossRef CAS PubMed .
-
(a) Y.-D. Lin, C.-T. Chien, S.-Y. Lin, H.-H. Chang, C.-Y. Liu and T. J. Chow, J. Photochem. Photobiol., A, 2011, 222, 192–202 CrossRef CAS ;
(b) P. Singh, A. Baheti, K. R. J. Thomas, C.-P. Lee and K.-C. Ho, Dyes Pigm., 2012, 95, 523–533 CrossRef CAS ;
(c) A. Venkateswararao, P. Tyagi, K. R. J. Thomas, P.-W. Chen and K.-C. Ho, Tetrahedron, 2014, 70, 6318–6327 CrossRef CAS .
-
(a) M. K. Nazeeruddin, P. Péchy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613–1624 CrossRef CAS PubMed ;
(b) M. K. Nazeeruddin, S. M. Zakeeruddin, R. Humphry-Baker, M. Jirousek, P. Liska, N. Vlachopoulos, V. hklover, C. H. Fisher and M. Grätzel, Inorg. Chem., 1999, 38, 6298–6305 CrossRef CAS PubMed .
-
(a) K. Sayama, S. Tsukagoshi, K. Hara, Y. Ohga, A. Shinpou, Y. Abe, S. Suga and H. Arakawa, J. Phys. Chem. B, 2002, 106, 1363–1371 CrossRef CAS ;
(b) G. Li, K.-J. Jiang, Y.-F. Li, S.-L. Li and L.-M. Yang, J. Phys. Chem. C, 2008, 112, 11591–11599 CrossRef CAS .
- B. Liu, Q. Liu, D. You, X. Li, Y. Naruta and W. Zhu, J. Mater. Chem., 2012, 22, 13348–13356 RSC .
-
(a) M. Pope and C. E. Swenberg, Electronic Processes in Organic Crystals and Polymers, Oxford University Press, Oxford, UK, 1999 Search PubMed ;
(b) W. Li, Y. Pan, L. Yao, H. Liu, S. Zhang, C. Wang, F. Shen, P. Lu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 892–901 CrossRef CAS .
-
(a) F. D. Lewis, R. S. Kalgutkar and J.-S. Yang, J. Am. Chem. Soc., 1999, 121, 12045–12053 CrossRef CAS ;
(b) J.-S. Yang, K.-L. Liau, C.-W. Tu and C.-Y. Hwang, J. Phys. Chem. A, 2005, 109, 6450–6456 CrossRef CAS PubMed .
-
(a) O. A. Kucherak, P. Didier, Y. Mély and A. S. Klymchenko, J. Phys. Chem. Lett., 2010, 1, 616–620 CrossRef CAS ;
(b) V. D. Gupta, V. S. Padalkar, K. R. Phatangare, V. S. Patil, P. G. Umape and N. Sekar, Dyes Pigm., 2011, 88, 378–384 CrossRef CAS ;
(c) E. L. Spitler, J. M. Monson and M. M. Haley, J. Org. Chem., 2008, 73, 2211–2223 CrossRef CAS PubMed .
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS .
-
(a) F. B. Dias, S. Pollock, G. Hedley, L.-O. Palsson, A. Monkman, I. I. Perepichka, I. F. Perepichka, M. Tavasli and M. R. Bryce, J. Phys. Chem. B, 2006, 110, 19329–19339 CrossRef CAS PubMed ;
(b) G. Krishnamoorthy and S. K. Dogra, J. Org. Chem., 1999, 64, 6566–6574 CrossRef CAS PubMed ;
(c) N. Mataga, H. Chosrowjan and S. Taniguchi, J. Photochem. Photobiol., C, 2005, 6, 37–79 CrossRef CAS ;
(d) E. Z. Von Lippert, Z. Naturforsch., A: Phys. Sci., 1955, 10, 541–545 Search PubMed ;
(e) N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1955, 28, 690–691 CrossRef CAS ;
(f) E. Z. VonLippert, Electrochem., 1957, 61, 962–975 Search PubMed .
-
(a) M. A. Webb, B. C. Morris, W. D. Edwards, A. Blumenfeld, X. Zhao and J. L. McHale, J. Phys. Chem. A, 2004, 108, 1515–1523 CrossRef CAS ;
(b) Y. I. Park, C.-Y. Kuo, J. S. Martinez, Y.-S. Park, O. Postupna, A. Zhugayevych, S. Kim, J. Park, S. Tretiak and H.-L. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 4685–4695 CrossRef CAS PubMed .
-
(a) M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed ;
(b) A. Hagfeldt and M. Grätzel, Chem. Rev., 1995, 95, 49–68 CrossRef CAS .
- B. Kaduk, T. Kowalczyk and T. V. Voorhis, Chem. Rev., 2011, 112, 321–370 CrossRef PubMed .
- R. G. Parr and W. Yang, Annu. Rev. Phys. Chem., 1995, 46, 701–728 CrossRef CAS PubMed .
- B. J. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, J. Phys. Chem. A, 2000, 104, 4811–4815 CrossRef CAS .
- A. D. Boese and J. M. L. Martin, J. Chem. Phys., 2004, 121, 3405–3416 CrossRef CAS PubMed .
-
(a) N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem., 1992, 70, 560–571 CrossRef CAS ;
(b) C. Sosa, J. Andzelm, B. C. Elkin, E. Wimme, K. D. Dobbs and D. A. Dixon, J. Phys. Chem., 1992, 96, 6630–6636 CrossRef CAS .
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed .
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed .
-
(a) S. R. Raga, E. M. Barea and F. Fabregat-Santiago, J. Phys. Chem. Lett., 2012, 3, 1629–1634 CrossRef CAS PubMed ;
(b) G. Li, M. Liang, H. Wang, Z. Sun, L. Wang, Z. Wang and S. Xue, Chem. Mater., 2013, 25, 1713–1722 CrossRef CAS .
- C.-H. Siu, L. Tien, L. Lee, P.-Y. Ho, P. Majumdar, C.-L. Ho, T. Chen, J. Zhao, H. Lie and W.-Y. Wong, J. Mater. Chem. C, 2014, 2, 7086–7095 RSC .
-
(a) W. H. Nguyen, C. D. Bailie, J. Burschka, T. Moehl, M. Grätzel, M. D. McGehee and A. Sellinger, Chem. Mater., 2013, 25, 1519–1525 CrossRef CAS ;
(b) W. Li, Y. Wu, X. Li, Y. Xie and W. Zhu, Energy Environ. Sci., 2011, 4, 1830–1837 RSC .
- M. Adachi, M. Sakamoto, J. Jiu, Y. Ogata and S. Isoda, J. Phys. Chem. B, 2006, 110, 13872–13880 CrossRef CAS PubMed .
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS .
-
(a) C. Kim, H. Choi, S. Kim, C. Baik, K. Song, M.-S. Kang, S. O. Kang and J. Ko, J. Org. Chem., 2008, 73, 7072–7079 CrossRef CAS PubMed ;
(b) H. Chen, H. Huang, X. Huang, J. N. Clifford, A. Forneli, E. Palomares, X. Zheng, L. Zheng, X. Wang, P. Shen, B. Zhao and S. Tan, J. Org. Chem., 2007, 72, 3652–3658 CrossRef PubMed .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed .
Footnote |
| † Electronic supplementary information (ESI) available: Absorption and emission spectra of dyes recorded in different solvents, CV recorded in DCM, NMR spectra (1H and 13C) and Cartesian coordinates of the dyes. See DOI: 10.1039/c4ra15234d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 




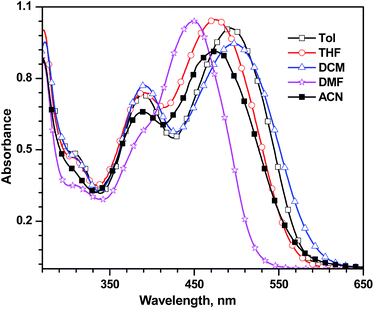
 .c Oxidation potentials are reported with reference to the ferrocene internal standard.d Deduced from the oxidation potential using the formula HOMO = 4.8 + Eox.e Deduced using the formula LUMO = HOMO − E0–0.f Calculated from intersection of absorption and emission spectra.g Calculated from E*ox = Eox − E0–0, with respect to NHE.h Reported for THF solutions.
.c Oxidation potentials are reported with reference to the ferrocene internal standard.d Deduced from the oxidation potential using the formula HOMO = 4.8 + Eox.e Deduced using the formula LUMO = HOMO − E0–0.f Calculated from intersection of absorption and emission spectra.g Calculated from E*ox = Eox − E0–0, with respect to NHE.h Reported for THF solutions.








