DOI:
10.1039/C4RA15207G
(Paper)
RSC Adv., 2015,
5, 3903-3907
Concentration dependent ratiometric turn-on selective fluorescence detection of picric acid in aqueous and non-aqueous media†
Received
25th November 2014
, Accepted 1st December 2014
First published on 1st December 2014
Abstract
A phosphorictriamide scaffold, [(NHAQ)3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O] (TAQP), which has an electron-rich aminoquinoline (AQ) chromophore, is shown to exhibit a ratiometric turn-on response for picric acid (PA) at low (1 × 10−5 M) concentration via a proton-transfer pathway. However, due to the presence of strong H-bonding and π⋯π interactions, at higher concentrations (1 × 10−3 M), it exhibits only a luminescence quenching behaviour for PA. These effects can be observed in both protic and non-protic systems. The detection limit for this aqueous phase turn-on sensing was found to be 0.2 ppm.
O] (TAQP), which has an electron-rich aminoquinoline (AQ) chromophore, is shown to exhibit a ratiometric turn-on response for picric acid (PA) at low (1 × 10−5 M) concentration via a proton-transfer pathway. However, due to the presence of strong H-bonding and π⋯π interactions, at higher concentrations (1 × 10−3 M), it exhibits only a luminescence quenching behaviour for PA. These effects can be observed in both protic and non-protic systems. The detection limit for this aqueous phase turn-on sensing was found to be 0.2 ppm.
Introduction
Detection of picric acid (PA) in water bodies is an important issue in society due to the increasing demands of national security and environmental protection.1 PA, one of the many commonly known hazardous nitro-explosives, is frequently found in soil and ground water in regions near armament facilities.2 Despite its higher explosive power than tri-nitrotoluene (TNT), PA is still being used in dye and leathering industries, firework and matchbox factories and in some pharmaceutical products.3 As a result, a vast amount of PA has been released into the environment. Due to its strongly acidic nature, it causes damage to skin, eye, liver and respiratory systems and it also causes a number of chronic diseases due to its slow degradation under physiological conditions.4 Several chromatographic and spectroscopic methods have been developed for PA sensing, of which fluorescence based techniques are more valuable due to their selectivity, sensitivity, cost efficiency and real-time detection.5 Over the years, a number of fluorescent materials have been reported based on small organic molecules, conjugated polymers and metal–organic frameworks that show a selective turn-off response towards PA.6 However, turn-off sensing methods are generally less desired as they offer a lower sensitivity and/or selectivity in the presence of other competing nitro-analytes. Thus, it is necessary to develop selective and sensitive turn-on sensors for nitro-explosives, although a few sensors based on turn-on responses have been reported for PA recently.7 Keeping this in mind, we have designed a new molecule on a phosphoramide tripodal backbone, [(NHAQ)3PO] (TAQP), which has an electron-rich aminoquinoline (AQ) chromophore. This compound, at low concentration (1 × 10−5 M), shows a selective ratiometric turn-on response towards PA, whereas at higher concentration (1 × 10−3 M) it shows a turn-off response for its excimer peak. Interestingly, the turn-on response for PA was observed for aqueous systems at a concentration of 1 × 10−5 M. To the best of our knowledge, this is the first report of concentration dependant turn-on and off type detection of PA in both protic and non-protic media.
Results and discussion
Crystal structure
The compound tris(3-aminoquinolino)phosphorictriamide (TAQP) was synthesized by the reaction of PCl5 with 3-aminoquinoline in toluene followed by hydrolysis and was characterized by 31P-NMR, mass-spectrometry and single-crystal X-ray diffraction (SC-XRD) (Scheme S1 and Fig. S1 and S2, ESI†). The SC-XRD analysis revealed that the compound was crystallized as TAQP·3H2O in the trigonal space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (Tables S1–S3, ESI†). The ligand core exhibits a C3 symmetric backbone along the axis of the PO bond. The phosphoryl oxygen atom (O1) is involved in a trifurcated H-bonding interaction with all of the three solvated water molecules (O1S–H2S) along the three-fold axis and these water molecules connect the adjacent N–H fragments to form a 1D-chain structure (Fig. 1(i) and (ii)). Interestingly, a hexagonal 3D-lattice (pseudo S6 symmetry) was obtained due to the O–H⋯N type H-bonding interaction of the remaining proton (H1S) of the water molecules with the N-donor sites (N13). In addition, TAQP·3H2O shows strong inter-sheet π⋯π stacking interactions between two adjacent AQ fragments (Fig. 1(iii)). The packing diagram of TAQP·3H2O shows a hexagonal open-channel structure, along the c-axis, with a void space of 201.3 Å3, which amounts to 14.3% of the unit-cell volume (Fig. 1(iv)).
(Tables S1–S3, ESI†). The ligand core exhibits a C3 symmetric backbone along the axis of the PO bond. The phosphoryl oxygen atom (O1) is involved in a trifurcated H-bonding interaction with all of the three solvated water molecules (O1S–H2S) along the three-fold axis and these water molecules connect the adjacent N–H fragments to form a 1D-chain structure (Fig. 1(i) and (ii)). Interestingly, a hexagonal 3D-lattice (pseudo S6 symmetry) was obtained due to the O–H⋯N type H-bonding interaction of the remaining proton (H1S) of the water molecules with the N-donor sites (N13). In addition, TAQP·3H2O shows strong inter-sheet π⋯π stacking interactions between two adjacent AQ fragments (Fig. 1(iii)). The packing diagram of TAQP·3H2O shows a hexagonal open-channel structure, along the c-axis, with a void space of 201.3 Å3, which amounts to 14.3% of the unit-cell volume (Fig. 1(iv)).
 |
| | Fig. 1 (i) Crystal structure of TAQP·3H2O at 50% ellipsoid probability; (ii) formation of a 1D-chain structure mediated by O–H⋯O interactions; (iii) and (iv) view of the hexagonal packing structure mediated by O–H⋯N and π⋯π interactions. | |
Photo-physical studies
The UV-Visible spectra of 1 × 10−5 M solutions of TAQP in both DMF and MeOH show an absorption maximum at 330 nm characteristic of the AQ chromophore (Fig. S3 and S4 ESI†). However, the fluorescence spectrum in MeOH shows two emission peaks at 367 and 405 nm (λex = 330 nm), while only one peak at 367 nm was observed in DMF. This indicates the possibility of H-bonded aggregation of TAQP in a protic solvent such as MeOH, even at concentrations as low as 1 × 10−5 M. Furthermore, the emission spectra of TAQP recorded in DMF–H2O (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) and MeOH–H2O (6:4) mixtures revealed the same trend, as two distinct emission peaks were obtained in both instances: 367 and 408 nm in DMF–H2O and 367 and 410 nm in MeOH–H2O.8 Noticeably, the peak around 405 nm was more prominent compared to the parent peak at 367 nm, suggestive of stronger H-bonding in the presence of water. In fact, in neat H2O suspensions the peak due to aggregation (at 419 nm) was alone prominent and the 367 nm emission was almost diminished. To further check this aggregation phenomenon, the emission spectra were recorded at higher concentrations, 1 × 10−4 M and 1 × 10−3 M, of TAQP. Interestingly, at the 1 × 10−3 M concentration of TAQP a large Stoke’s shifted (100 nm) peak at 470 nm was observed in both DMF and MeOH in addition to weak emission signals at 367 and 405 nm. This new red shifted peak is attributed to the formation of an excimer complex at 1 × 10−3 M, which is absent at both 1 × 10−4 M and 1 × 10−5 M concentrations of TAQP (Fig. 2 and S5–S8, ESI†).9 Formation of this excimer emission can be attributed to a strong aggregation of the TAQP molecules at this concentration, mediated by the rich H-bonding and π⋯π interactions as observed from its crystal structure (Fig. 1(iv)).
:4) and MeOH–H2O (6:4) mixtures revealed the same trend, as two distinct emission peaks were obtained in both instances: 367 and 408 nm in DMF–H2O and 367 and 410 nm in MeOH–H2O.8 Noticeably, the peak around 405 nm was more prominent compared to the parent peak at 367 nm, suggestive of stronger H-bonding in the presence of water. In fact, in neat H2O suspensions the peak due to aggregation (at 419 nm) was alone prominent and the 367 nm emission was almost diminished. To further check this aggregation phenomenon, the emission spectra were recorded at higher concentrations, 1 × 10−4 M and 1 × 10−3 M, of TAQP. Interestingly, at the 1 × 10−3 M concentration of TAQP a large Stoke’s shifted (100 nm) peak at 470 nm was observed in both DMF and MeOH in addition to weak emission signals at 367 and 405 nm. This new red shifted peak is attributed to the formation of an excimer complex at 1 × 10−3 M, which is absent at both 1 × 10−4 M and 1 × 10−5 M concentrations of TAQP (Fig. 2 and S5–S8, ESI†).9 Formation of this excimer emission can be attributed to a strong aggregation of the TAQP molecules at this concentration, mediated by the rich H-bonding and π⋯π interactions as observed from its crystal structure (Fig. 1(iv)).
 |
| | Fig. 2 Emission spectra of TAQP (a) in neat DMF and (b) in protic environment (λex = 330 nm); insets show the emission colours under UV-lamp (λex = 365 nm) in (i) 1 × 10−5 M solution of DMF, (ii) 1 × 10−3 M solution of DMF and (iii) 1 × 10−5 M solution of DMF–H2O or MeOH–H2O. | |
To further confirm this aggregation phenomenon, field emission scanning electron microscopy (FESEM) images were recorded for TAQP films prepared from both non-aqueous and aqueous media at each of the concentrations. The FESEM images of the TAQP samples prepared from 1 × 10−5 M solutions indicate that the aggregation is dominant in aqueous systems (MeOH–H2O (6:4) or DMF–H2O (6:4)) compared to those prepared in neat DMF. However at higher (1 × 10−3 M) concentrations, strong aggregation can be seen in both protic and aprotic media, forming uniform spherical particles about 800 nm in size. These observations strongly suggest that non-covalent interactions such as H-bonding and π⋯π interactions are prevalent at higher solution concentrations of TAQP (Fig. 3 and S9, ESI†). The fluorescence decay measurements in neat DMF reveal that the lifetime of the excimer peak at 467 nm in the higher concentration is longer (τ1 = 4.25 and τ2 = 12.31 ns) compared to that of the 367 nm peak (τ1 = 1.09 and τ2 = 4.47 ns) in the lower concentration (Fig. S10, ESI†).
 |
| | Fig. 3 FESEM images of 1 × 10−5 M solutions of TAQP in MeOH (a) before and (c) after the addition of PA; FESEM images of 1 × 10−3 M solutions of TAQP in DMF (b) before and (d) after the addition of PA. | |
Picric acid sensing
Prompted by the rich photophysical behaviour of TAQP and the presence of multiple basic N-donor sites in its structure, we were interested in exploring its potential as a PA sensor in both 1 × 10−5 M and 1 × 10−3 M solution concentrations. Upon the titration of the 1 × 10−5 M solution of TAQP in DMF, the 367 nm emission gradually decreased and a new peak at 465 nm was observed after the addition of 2.5 equivalents of PA. At a concentration of 5 equivalents of PA, the peak at 367 nm is almost diminished as the new peak at 465 nm becomes dominant, signifying a ratiometric fluorescence response. Similar behaviour was observed when the titration was performed in MeOH; the peaks at 367 and 405 nm were quenched with the onset of the new peak at 465 nm (Fig. 4 and S11 and S12, ESI†).
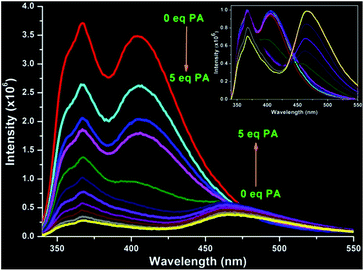 |
| | Fig. 4 Emission spectrum of TAQP in MeOH with addition of 5 equivalents of PA. Inset: normalised spectrum showing the new emission peak at 465 nm. | |
Time decay measurements at this new peak gave higher life time values of τ1 = 3.88 and τ2 = 10.62 ns, indicating a stronger interaction between TAQP and PA (Fig. S10, ESI†). Concurrently, the 330 nm peak in the UV-visible spectrum of TAQP (in DMF or in MeOH) was found to be gradually red-shifted to an intense 360 nm peak with the incremental addition of PA (Fig. S13 and S14, ESI†). These observations point to the fact that the origin of the new peak at 465 nm can be attributed to the protonation of the N-donor sites of TAQP by PA.
To validate this, emission spectral titrations of TAQP were performed in the presence of trifluoroacetic acid (TFA), a comparably strong acid compared with PA.7b These again showed a ratiometric turn-on response, similar to that of PA, upon the addition of 5 equivalents of TFA. Further, the 1H-NMR spectra of TAQP in the presence of 10 equivalents of TFA showed a downfield shift for the AQ protons. A similar downfield shift was observed for the TAQP sample containing 5 equivalents of PA as well. Thus, both these results strongly suggest that TAQP exhibits a ratiometric fluorescence response due to proton transfer from the acidic PA to the N-donor sites of the AQ moieties (Fig. 5 and S15–S17, ESI†). Also, it is interesting to note that TFA shows only a ratiometric fluorescence response, whereas PA largely quenched the emission due to the AQ chromophore. This is due π⋯π interactions between PA and the quinoline moiety, which results in an effective energy transfer from AQ to PA. This interaction was clearly visible in the FESEM images, in which a better and different kind of aggregation was observed for TAQP (1 × 10−5 M) treated with 5 equivalents of PA (Fig. 3). Also, titration experiments were performed in aqueous systems containing DMF–H2O (6:4) or MeOH–H2O (6:4), which again gave the ratiometric response with the formation of a peak at 465 nm (Fig. S18–S20, ESI†). Furthermore, titration experiments with PA in neat H2O suspensions (1 × 10−5 M) showed a facile ratiometric response wherein the 419 nm aggregation peak was completely shifted to a new peak due to protonation at 464 nm (Fig. 6 and S21, ESI†). The detection limits for this aqueous phase sensing in neat H2O and DMF–H2O mixture were found to be 0.2 and 0.13 ppm, respectively (Fig. S22, ESI†).7a
 |
| | Fig. 5 1H-NMR of TAQP (brown) in the presence of PA (green) and TFA (blue) in DMSO-d6. The signals marked in boxes are the aromatic protons adjacent to the N-donor sites. | |
 |
| | Fig. 6 Relative changes of fluorescence intensity (I464/I419) of TAQP (1 × 10−5 M) with PA in neat H2O suspension. | |
Similar PA titrations with the 1 × 10−3 M TAQP solution show only a quenching behaviour. Thus, a gradual decrease in the excimer 465 nm peak was observed with the incremental addition of PA, with no apparent shift in its emission maxima. This observation suggests that the acidic protons of PA could not disrupt the strong inter-molecular H-bonding present in the higher concentrations of TAQP and thus could not protonate the N-donor sites present in it. However, PA can interact with the surface aromatic groups of the AQ moieties via π⋯π interactions and can quench its luminescence emission.6e This is in fact evident from the FESEM image of the TAQP (1 × 10−3 M) treated with PA (20 eq.), which showed that the hollow spherical aggregation was retained as these particles were merely covered with the PA layers (Fig. 3, S23 and S24, ESI†).
To check the selectivity of TAQP towards PA, titration experiments were performed for other nitro-analytes such as nitrobenzene (NB), dinitrobenzene (DNB), nitrotoluene (NT), dinitrotoluene (DNT), trinitrotoluene (TNT), cyclotrimethylenetrinitramine (RDX) and nitrophenol (NP). These titrations show negligible luminescence quenching for most of the analytes, except for NP. For NP only a moderate quenching behaviour and no ratiometric/turn-on response was observed with a 1 × 10−5 M TAQP solution (Fig. 7 and S25–S33, ESI†).
 |
| | Fig. 7 (a) Relative changes in fluorescence intensity (I465/I367) of TAQP (1 × 10−5 M) for PA and other possible interfering analytes. (b)–(e) Emission colours under a UV-lamp for TLC plates treated with TAQP and TAQP + PA at different concentrations. | |
Furthermore, the selectivity of TAQP towards PA in the presence of other interfering analytes has been determined from luminescence titration with mixed nitro-analytes (Fig. 8). It is evident from these titrations that only PA can show the ratiometric response, while the other analytes exhibit negligible changes in emission. Furthermore, the obtained Stern–Volmer constant (KSV) value of 2.8 × 105 M−1 for this ratiometric sensing is quite high and is comparable with other recently reported PA sensors.10 The sensing selectivity of TAQP for PA in the presence of other interfering aromatic functionalities such as 4-bromobenzoic acid (4-BBA), 4-nitrobenzoic acid (4-NBA), benzaldehyde (Bz), 4-nitrobenzaldehyde (4-NBz) and 4-naphthol (4-NpOH) have also been tested, which again showed the ratiometric response for only PA (Fig. S34, ESI†).
 |
| | Fig. 8 Emission spectral titration of TAQP (1 × 10−5 M) of PA (1 × 10−3 M) in the presence of other interfering nitro-analytes (1 × 10−3 M) in DMF. | |
Conclusions
In summary, we present a simple aminoquinoline containing tripodal phosphoramide ligand that shows concentration dependant aggregation due to H-bonding and π⋯π stacking in both protic and non-protic media. At a 1 × 10−5 M concentration, a ratiometric turn-on response was observed for PA with the formation of a new peak at 465 nm due to protonation of its N-donor sites. Furthermore, TAQP shows a high selectivity in ppm levels towards PA in aqueous media, as well as in the presence of other interfering nitro-explosives, and has the potential to act as a PA probe for real-time applications.
Experimental section
General remarks
All manipulations involving phosphorus halides were performed under dry atmosphere in standard Schlenk-glassware. 3-Aminoquinoline was purchased from Aldrich and used as received. PCl5 and POCl3 were purchased locally (SPECTROCHEM, India) and used as received. NMR spectra were recorded on a 400 MHz Jeol FT spectrometer (1H-NMR: 400.13 MHz, 13C{1H}-NMR: 100.62 MHz, 31P{1H}-NMR: 161.97 MHz) at room temperature using SiMe4 (1H, 13C) and 85% H3PO4 (31P) as external standards. Thermal analysis data has been obtained from a Perkin-Elmer STA-6000 thermogravimetric analyser. Elemental analyses were performed on a Vario-EL cube elemental analyser. FT-IR spectra were taken on a Perkin-Elmer spectrophotometer. The absorption and emission studies were done by a Perkin-Elmer Lambda 45 UV-Visible spectrophotometer and SPEX Flurolog HORIBA JOBIN VYON fluorescence spectrophotometer with a double-grating 0.22 m SPEX 1680 monochromator and a 450 W Xe lamp as the excitation source. The excitation and emission spectra of the complexes were corrected at instrumental function. The photoluminescence lifetime measurements were carried out using a SPEX Flurolog HORIBA JOBIN VYON 1934 D phosphorimeter. The luminescent lifetime of TAQP was measured using a Time Correlated Single Photon Counting (TCSPC) method at 298 K. The 298 K luminescence decay profiles were fitted to biexponential curves. Similarly, the lifetimes were fitted by using the DAS software.
Synthesis of TAQP
To a stirred solution of 3-aminoquinoline (6.92 g, 0.048 mol) in toluene (150 mL), PCl5 (1 g, 0.0048 mol) in toluene (50 mL) was added drop wise for 30 minutes. The resulting mixture was refluxed at 110 °C for 12 h to yield a yellow precipitate. The precipitate was collected by filtration and washed with excess of water to remove the amine-hydrochloride by-product. The resulting powder was treated with NaOH solution in methanol and washed with water. The obtained yellow powder was dried in a vacuum and collected. Crystals suitable for X-ray analysis were obtained from its solution in methanol. Yield: 66% (1.20 g based on P). 1H-NMR (400 MHz, (CD3)2SO: δ 7.56 (dd, 3H, CH), 7.58 (dd, 3H, CH), 7.85 (d, 3H, CH), 7.92 (d, 3H, CH), 8.13 (s, 3H, CH), 8.87 (s, 3H, CH), 8.99 (d, 3H, NH). 31P NMR (161 MHz, {(CD3)2SO}): δ −4.02. FT-IR data in KBr pellet (cm−1): 3100, 1605, 1575, 1511, 1457, 1410, 1346, 1207, 1184, 1139, 1105, 993, 940, 891, 865, 620, 569. Anal. calcd for C27H21N6OP: C, 68.06; H, 4.44; N, 17.64. Found: C, 67.96; H, 4.56; N, 16.61.
Crystallography
Reflections of TAQP·3H2O were collected on a Bruker Smart Apex Duo diffractometer at 100 K using MoKα radiation (λ = 0.71073 Å) (Table S1, ESI†). Structures were refined by full-matrix least-squares against F2 using all data (SHELX97).11 All non-hydrogen atoms were refined anisotropically if not stated otherwise. Hydrogen atoms were constrained in geometric positions to their parent atoms. Crystals of TAQP·3H2O diffracted weakly at higher angles and hence a 2θ = 50° cut-off was applied.
Acknowledgements
We thank the DST, India (SR/S1/IC-50/2012 (R.B.) and IISER, Pune for financial support. AY thanks CSIR, India for a research fellowship. We thank HEMRL, Pune for providing TNT and RDX samples to IISER, Pune.
Notes and references
-
(a) S. W. Thomas III, G. D. Joly and T. M. Swager, Chem. Soc. Rev., 2007, 36, 1339 CrossRef PubMed;
(b) M. E. Germain and M. J. Knapp, Chem. Soc. Rev., 2009, 38, 2543 RSC.
- H. Muthurajan, R. Sivabalan, M. B. Talawar and S. N. Asthana, J. Hazard. Mater., 2004, 112, 17 CrossRef CAS PubMed.
-
(a) E. H. Volwiler, Ind. Eng. Chem., 1926, 18, 1336 CrossRef CAS;
(b) D. T. Meredith and C. O. Lee, J. Am. Pharm. Assoc., 1939, 28, 369 CrossRef CAS.
-
(a) Safety Data Sheet for Picric Acid, Resource of UK National Institute of Health;
(b) P. C. Ashbrook and T. A. Houts, ACS Div. Chem. Health Safety, 2003, 10, 27 Search PubMed.
-
(a) G. He, H. Peng, T. Liu, M. Yang, Y. Zhang and Y. Fang, J. Mater. Chem., 2009, 19, 7347 RSC;
(b) Y. Salinas, R. Martínez-Máñez, M. D. Marcos, F. Sancenón, A. M. Costero, M. Parra and S. Gil, Chem. Soc. Rev., 2012, 41, 1261 RSC.
-
(a) M. E. Germain and M. J. Knapp, Chem. Soc. Rev., 2009, 38, 2543 RSC;
(b) T. H. Liu, L. P. Ding, K. R. Zhao, W. L. Wang and Y. Fang, J. Mater. Chem., 2012, 22, 1069 RSC;
(c) V. Bhalla, A. Gupta and M. Kumar, Org. Lett., 2012, 14, 3112 CrossRef CAS PubMed;
(d) B. Roy, A. K. Bar, B. Gole and P. S. Mukherjee, J. Org. Chem., 2013, 78, 1306 CrossRef CAS PubMed;
(e) V. Bhalla, A. Gupta, M. Kumar, D. S. S. Rao and S. K. Prasad, ACS Appl. Mater. Interfaces, 2013, 5, 672 CrossRef CAS PubMed;
(f) Y. Peng, A.-J. Zhang, M. Dong and Y.-W. Wang, Chem. Commun., 2011, 47, 4505 RSC;
(g) S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 2881 CrossRef CAS PubMed;
(h) S. J. Toal and W. C. Trogler, J. Mater. Chem., 2006, 16, 2871 RSC.
-
(a) Y. Xu, B. Li, W. Li, J. Zhao, S. Sun and Y. Pang, Chem. Commun., 2013, 49, 4764 RSC;
(b) S. Kaur, V. Bhalla, V. Vij and M. Kumar, J. Mater. Chem. C, 2014, 2, 3936 RSC;
(c) J. C. Sanchez and W. C. Trogler, J. Mater. Chem., 2008, 18, 3143–3156 RSC.
-
(a) U. Rosch, S. Yao, R. Wortmann and F. Wurthner, Angew. Chem., Int. Ed., 2006, 45, 7026 CrossRef PubMed;
(b) H. Yao, K. Domoto, T. Isohashi and K. Kimura, Langmuir, 2005, 21, 1067 CrossRef CAS PubMed.
- R. P. Balaunstein and K. S. Gan, Photochem. Photobiol., 1973, 18, 347 CrossRef PubMed.
- D. J. Desilets, P. T. Kissinger and F. E. Lytle, Anal. Chem., 1987, 59, 1244 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional figures and tables. CCDC 1024970. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra15207g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 







