
Revealing the properties of the cubic ZrO2 (111) surface by periodic DFT calculations: reducibility and stabilization through doping with aliovalent Y2O3
Abstract
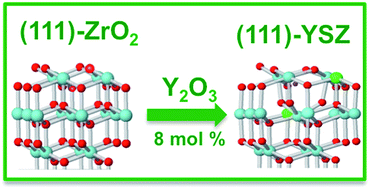
A detailed theoretical study concerning the formation of oxygen vacancies on the clean (111) surface of cubic ZrO2 and the structural and electronic properties of the (111) surface of yttria-stabilized zirconia (YSZ, 8 mol% Y2O3) was carried out using DFT methods in a periodic approach. For the formation of oxygen defects on the clean (111) surface, two different oxygen vacancy positions and two possible spin states for each position were investigated. Large vacancy formation energy, small relaxation and the presence of a highly localized state in the gap characterize the formation of oxygen defects on this surface. Regarding the yttria-stabilized surface, a systematic study of the stability, geometry and electronic structure of seven different configurations for Y atoms and oxygen vacancies on the surface was performed. The doping with Y2O3 stabilizes the cubic (111) ZrO2 surface and is accompanied by large relaxations of the O atoms NN to the vacancies. In addition, Y atoms preferentially occupy positions NNN to the defect. Despite the presence of 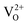 vacancies in YSZ, no mid-gap states have been observed in any of the studied arrangements. This study allowed identifying an accurate computational protocol and a suitable model of the (111) surface of YSZ, through the characterization of its structural and electronic properties. Both could be used to further elucidate the role of YSZ as electrolyte in SOFC applications, with a view to better clarifying the basic operating principles of low temperature solid oxide fuel cells (LT-SOFCs).
vacancies in YSZ, no mid-gap states have been observed in any of the studied arrangements. This study allowed identifying an accurate computational protocol and a suitable model of the (111) surface of YSZ, through the characterization of its structural and electronic properties. Both could be used to further elucidate the role of YSZ as electrolyte in SOFC applications, with a view to better clarifying the basic operating principles of low temperature solid oxide fuel cells (LT-SOFCs).
 Please wait while we load your content...
Please wait while we load your content...

