
Influence of less active initiator on the living performance of atom transfer radical polymerization and the structure of the synthesized grafted copolymer†
Abstract
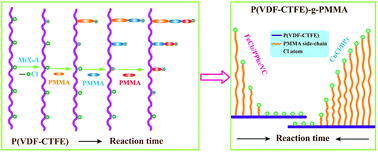
In an effort to precisely describe the structure of polymers synthesized via ATRP initiated with less active initiator and clearly understand the evolution of the forming polymer chains, methyl methacrylate (MMA) was grafted onto poly(vinylidene fluoride-chlorotrifluoroethylene) (P(VDF-CTFE)) using two different catalysts. The detailed structural information of the grafted copolymers, including the grafting density, the average side chain length, and the average molecular weight and its distribution, has been carefully determined by removing the homopolymers from the crude copolymer and converting any uninitiated Cl atoms into H atoms, which has been finely correlated to the reaction conditions. By correlating the structural information to a kinetic analysis, the step-by-step evolution of the side chains has been clearly demonstrated. The low initiation activity of C–Cl and the low redox capability of the catalyst system were found to be responsible for the wide molecular weight distribution, the low grafting density and the slow growing polymer chains. The results may help to understand not only the polymer structure synthesized from the ATRP process more precisely but also the evolution of the polymer chains under different reaction conditions.
 Please wait while we load your content...
Please wait while we load your content...

