
Ruthenium complexes as inhibitors of human islet amyloid polypeptide aggregation, an effect that prevents beta cell apoptosis†
Abstract
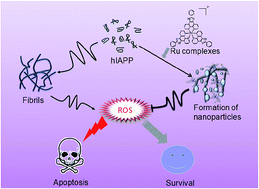
Human islet amyloid polypeptide (hIAPP) aggregation is essential in the loss of insulin-producing pancreatic beta cells in type 2 diabetes mellitus (T2DM). Recent studies have identified hIAPP fibril as a therapeutic target of T2DM. Metal complexes could covalently bind to the intracellular peptides to regulate their biological functions. In the present study, ruthenium (Ru) complexes NAMI-A (1) [Ru(bpy)3](ClO4)2 (2) (bpy = 2,2′-dipyridyl), [Ru(pip)3](ClO4)2 (3) (pip = 2-phenylimidazo[4,5-f]-[1,10]phenanthroline) and [Ru(phtpy)(phen)Cl]ClO4 (4) (phtpy = 2,6-bis(2-pyridyl)-4-phenylpyridine, phen = 1,10-phenanthroline) were selected to investigate their influence on hIAPP fibrillation in vitro. The results of thioflavin T (ThT) fluorescence assay showed that Ru complexes effectively inhibited the formation of hIAPP fibril. AFM images and TEM images further validated that the hIAPP fibrillation was disaggregated by the Ru complexes and then formed nanoscale particles, which tends to be a time-dependent process. Moreover, Ru complexes demonstrated a protective effect towards hIAPP-caused cell damage by restraining ROS generation and blocking cell apoptosis. In addition, it has been found that Ru complexes can also disaggregate hIAPP fibrils effectively inside the cells, and that the effects were proportional to the lipophilicity of the Ru complexes. Taken together, this study provides a strategy for designing Ru complexes for treating T2DM by targeting hIAPP.
- This article is part of the themed collection: Towards understanding and treating Alzheimer’s disease
 Please wait while we load your content...
Please wait while we load your content...

