DOI:
10.1039/C4RA15147J
(Paper)
RSC Adv., 2015,
5, 23548-23555
Coaxial carbon nanofiber/NiO core–shell nanocables as anodes for lithium ion batteries
Received
24th November 2014
, Accepted 10th February 2015
First published on 11th February 2015
Abstract
Hierarchically coaxial carbon nanofiber/NiO (CNF/NiO) core–shell nanocables for lithium ion batteries are prepared to coat α-Ni(OH)2 on the surface of electrospun carbon nanofibers (CNF) by electrophoretic deposition, followed by thermal processing in air. In the coaxial CNF/NiO nanocables, a NiO shell of about 20 nm thick is formed by coating with nano-furs outward on the surface of a CNF core of 200 nm in diameter, which is the main factor for providing a three-dimensional (3D) structure. The NiO shells, comprising of abundant inner spaces on the surface of CNF and high conductivity of 1D CNF, are deeply dependent on the enhancement of electrochemical rate capability. Abundant inner spaces in the NiO shell and the interconnected network between nanocables facilitate the mass transfer. The CNF core with the cushioning effect created through the elastic deformation provides electrochemical stability by protecting both radial compression and volume expansion originating from NiO shells radially. The CNF/NiO nanocables deliver a high reversible capacity of 825 mA h g−1 at 200 mA g−1 after 50 charge–discharge cycles without showing obvious decay. The coaxial CNF/NiO nanocables increase not only electrochemical capability but also electrochemical stability.
1. Introduction
Electrically active transition metal oxides (MxOy, M = Ni, Co, Cu, Fe, Mn) have attracted much attention and widespread interest in the energy industry, due to their high theoretical capacity on the basis of their unique conversion mechanism, (MO + 2Li+ + 2e− = Li2O + M), long cycle life and high recharging rates.1–8 Such desirable properties enable them to make promising anode materials in lithium ion batteries (LIBs). Among them, nickel oxide (NiO) has been extensively investigated as an important functional inorganic material, for use in fuel cells, solar cells, LIBs and supercapacitors, owing to its superior properties such as its high theoretical capacity (718 mA h g−1), higher density (6.81 g cm−3) than that of graphite (2.268 g cm−3), nontoxicity and low material cost.9–15 Unfortunately, pure transition metal oxides such as NiO give rise to poor cycling performance during cycling, owing to the tendency of particle agglomeration during lithiation/delithiation processes and mechanical instabilities caused by drastic volume changes, which ultimately result in increased diffusion lengths and electrical disconnection from the current collector.16–19 Specifically, as anodic materials, the electrochemical processes of NiO can be revealed as not an intercalation but a conversion process: NiO + 2Li+ + 2e− = Li2O + Ni. This leads to the formation of metallic Ni and Li2O nanoparticles accompanied with large volume expansion. Therefore, NiO electrodes suffer severe mechanical disintegration due to the drastic volumetric changes during lithiation and delithiation, and this leads to rapid deterioration in capacity. Accordingly, much research involving NiO has focussed on increasing the specific capacity, long-term cyclic performance, and reversible capacity by preparing NiO/graphene composites, NiO/MWCNT composites, NiO/carbon core–shell nanowire array.20–23 Nevertheless, it is hard to manipulate the capacity decay by lithiated NiO volume expansion.
Electrophoretic deposition (EPD) is a reliable method that can coat α-Ni(OH)2 nanoparticles from a Ni(NO3)2 solution on the surface of a CNF cathode under an applied electric field.24–26 This useful technique is remarkably unique and so novel that it has not been used for a CNF/NiO system previously. Under the influence of an electric field, charged ions in a solution move toward the oppositely charged electrode by electrophoresis. After the charged ions accumulate at the electrode, they deposit easily as proper structures according to the rate of mass transfer with applied voltage. The deposited materials are crystallized through a thermal process. The CNF/NiO nanocables to be designed by an EPD method possess a 3D hierarchically porous structure, which originates from abundant inner spaces on the surface of CNF and high conductivity of 1D CNF and the interconnected network between nanocables. The NiO shells with abundant inner spaces in their 3D hierarchical network with coaxial CNF/NiO core–shell structure leads to excellent rate capability. The abundant inner spaces in NiO shells enables the electrolyte to access easily the NiO anode material. However, in order to maintain this rate capability, the volume expansion along with radial compression by lithiated NiO should be effectively avoided. NiO has characteristics of inelastic deformation, while CNF is known to show elastic deformation with high elastic modulus. The CNF core plays an important role in protecting volume expansion along with radial compression of the lithiated NiO shell during cycling by creating a cushioning effect.27,28
As shown in recent research, the performances of CNF/Ni,29 PAN/PPy-based CNF,30 and porous NiO,31 are still lower than those of CNF/NiO core–shell nanocables. For CNF/Ni, the CNF/Ni shows a low capacity because the Ni particles of CNF/Ni are too deeply buried to the extent that Ni particles cannot react with lithium ions. The PAN/PPy-based CNF, which is carbonized by bicomponent polymer solution represents low performance because there is no NiO shell structure, although it offers the suitable sites to store lithium from the point of view carbon structure. The porous NiO without support material such as CNF illustrates low performance due to its pulverized characteristics, gives rise to fading of the capacity caused by loss of electron pathway. However, the CNF/NiO core–shell nanocables offer not only electrochemical rate capability but also electrochemical stability, because of its high conductivity and 1D conductive pathway with minimized resistance.
An aim in this study is to prepare a novel 3D coaxial CNF/NiO nanocable to have both high rate capability and excellent electrochemical stability at the same time. The CNF/NiO nanocables are prepared by directly coating with α-Ni(OH)2 nanoparticles on CNF through an electrophoretic deposition (EPD), followed by a thermal process.
2. Results and discussion
The process of α-Ni(OH)2 deposition on the surface of CNF through an electrophoretic deposition (EPD) technique is shown in Fig. 1. Under an applied electrical field, the Ni2+ ions in a Ni(NO3)2·6H2O ethanol solution move toward the surface of one-dimensional (1D) CNFs as a cathode, and then positively charged CNF–Ni2+ is formed, with the Ni2+ ions adsorbed on the CNF. At the same time, NO3− ions of Ni(NO3)2 are electrochemically reduced with H2O, and then the produced OH− ions move toward CNF–Ni2+ without diffusing into the bulk solution. Afterward, α-Ni(OH)2 on the surface of CNF is formed by rapidly reacting Ni2+ with OH−. After calcining at 300 °C for 2 h, the CNF/α-Ni(OH)2 is converted into crystallized CNF/NiO. The mechanism that NiO is formed on the surface of CNF is as follows:| | |
CNF (cathode) + Ni2+ = CNF–Ni2+ (adsorption)
| (1) |
| | |
NO3− + H2O + 2e− = NO2− + 2OH−
| (2) |
| | |
2H2O + 2e− = 2OH− + H2
| (3) |
| | |
CNF–Ni2+ + 2OH− = CNF/α-Ni(OH)2
| (4) |
| | |
CNF/α-Ni(OH)2 → CNF/NiO (calcination)
| (5) |
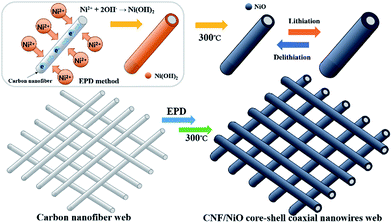 |
| | Fig. 1 Fabricating process of CNF/NiO. | |
The CNF/NiO nanocables prepared by an EPD technique offer not only a 3D hierarchically porous core–shell structure, but also important characteristics such as mechanical flexibility, compressibility, mechanical stability capable of withstanding during cycling, and excellent cohesion between the inelastic NiO shell and the elastic CNFs core.
Fig. 2 shows the SEM images for the surface of NiO powder, pure CNF, and CNF/NiO. In Fig. 2a and b, NiO powders represent the rectangular-like shape, in which the agglomerated particles range in size from 200 nm to 500 nm. In Fig. 2c and d, pure CNFs display the woven network structure , which is partially aligned along the winding direction of the drum winder. The CNFs have a high elastic modulus and can be restored against a compressive load. Highly conductive 1D woven network structures can be well matched with other functional materials to form novel structures for emerging applications. As for Fig. 2e and f, the CNF/NiO was prepared by coating α-Ni(OH)2 on the surface of CNFs through an EPD process with a weak applied voltage of 10 V for 3 h, and subsequent heat treatment at 300 °C for 2 h. The NiO on the surfaces of the CNFs is deposited uniformly throughout the woven network CNF. In Fig. 2e, the NiO nanoparticles appear to be coated on the CNF core. Analyzing the magnified Fig. 2f more accurately, the CNF/NiO nanocables have a 3D coaxial structure coated with NiO shell on the surface of the CNF core, showing that the diameters of core and shell are 110 nm and 220 nm, respectively. The NiO shell stores lithium ion compactly. The 3D porous structure of CNF/NiO nanocables produced by the interlayers of CNFs as woven networks (Fig. 2e) leads to abundant inner space between CNFs, offering tremendous channels for facile electrolyte flow, and inducing excellent contact between the electrolyte and NiO. These characteristics facilitate mass transfer and charge transfer in enhancing the electrochemical performance.
 |
| | Fig. 2 SEM images for (a) and (b) NiO, (c) and (d) CNF, and (e) and (f) CNF/NiO. | |
Fig. 3 shows TEM images for NiO powder, and CNF/NiO. In Fig. 3a and b, NiO powders show a polygonal shape with strong agglomeration. Furthermore, the particle size of the synthesized NiO varies from 20 to 100 nm. The CNF/NiO as shown in Fig. 3c consists of a CNF core and NiO shell with 3D coaxial morphology. The NiO coating around the CNF is certainly uniform, with a thickness of around 20 nm (Fig. 3c). The CNF core of diameter 200 nm, which acts as the electrical pathway for the coaxial structure, is seen clearly. Fig. 3d represents the HR-TEM image of the NiO shell. The NiO shell consists of many nanoparticles, and the size of these nanoparticles is in the range of 3 to 5 nm in diameter. The lattice spacing (d = 2.08 Å) between the lattice fringes agrees with orientation (2 0 0) plane, as observed from the analysis of XRD (Fig. 4).32 For the CNF/NiO nanocable with core–shell structure, the NiO shell has the characteristic of inelastic deformation, whereas the CNF is known as elastic deformation. During lithiation, NiO in the shell is compressed in the radial direction through inelastic flow and volume expansion of the NiO shell is mostly in the radial direction. Even if the lithiated NiO shell is enlarged during cycling, the elasticity of CNF with high modulus enables to protect the battery failure from NiO inelastic flow caused by volume variation.
 |
| | Fig. 3 TEM images for (a) and (b) NiO and (c) and (d) CNF/NiO. | |
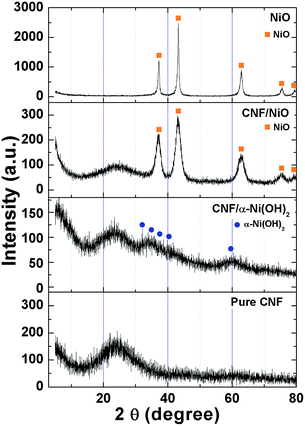 |
| | Fig. 4 XRD patterns of CNF, CNF/α-Ni(OH)2, NiO and CNF/NiO. | |
Fig. 4 represents the XRD patterns of NiO powder, pure CNF, CNF/α-Ni(OH)2, and CNF/NiO. The CNF exhibits a broad peak around 24°, indicating the typical amorphous structure. The diffraction peaks of the CNF/α-Ni(OH)2 can be indexed to hexagonal nickel hydroxide hydrate (α-3Ni(OH)2·H2O, JCPDS card no. 22-0444). The major diffraction peaks of NiO powder is formed at 2θ = 37.2°, 43.2°, 62.8°,75.3° and 79.3°, corresponding to (1 1 1), (2 0 0), (2 2 0), (3 1 1) and (2 2 2) planes of the cubic NiO phase (JCPDS card no. 04-0835), respectively. The peaks of CNF/NiO almost coincide with those of pure NiO particles, indicating that α-Ni(OH)2 adsorbed on the CNF is well transformed with CNF/NiO coaxial nanocables. The diffraction peaks of CNF/NiO are weaker and wider than those of NiO powder. This evidence indicates that α-Ni(OH)2 is slowly deposited as the nano-sized particles on the surface of CNF caused by the effect of slow mass transfer of 10 V DC in EPD process, along with the subsequent formation of CNF/NiO coaxial nanocables by annealing of 300 °C for 2 h.
Fig. 5 shows the TGA results of NiO powder, pure CNF, and CNF/NiO in air. The NiO powder shows a weight loss of 9.3 wt% due to water evaporation. Pure CNF shows the weight loss by water evaporation in the rage of 25 to 200 °C, and is decomposed completely at about 630 °C. For CNF/NiO, the degradation of three steps is observed. The first weight loss is due to water evaporation and solvent in the range of 25 to 200 °C. The second weight loss was due to the degradation of CNF side chain between 200 and 360 °C. The last step occurs in the range of 360 to 630 °C because of the complete decomposition of CNF main chain, representing that NiO content remains 54.8 wt% in CNF/NiO. This means that the weight ratio of NiO to CNF is 54.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45.2. This ratio is used in calculating the theoretical capacity of CNF/NiO nanocomposite.
:45.2. This ratio is used in calculating the theoretical capacity of CNF/NiO nanocomposite.
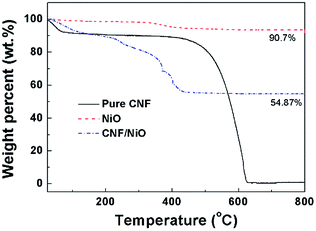 |
| | Fig. 5 TGA thermograms of CNF, NiO, and CNF/NiO in air. | |
Fig. 6 shows the initial and second charge and discharge profile for NiO powder, and CNF/NiO anodes with the voltage ranging from 0.02–3 V at a current density of 200 mA g−1. For pure CNF, shown in Fig. 6a, the specific capacity is much lower than for other two samples. For NiO powder, shown in Fig. 6b, the initial charge and discharge capacities of the NiO powder are 1370 and 813 mA h g−1, respectively, and the second charge and discharge capacities 845 and 765 mA h g−1, respectively. The NiO electrode represents high irreversible capacity during the initial cycle, followed by abrupt capacity decay after the second cycle compared to CNF/NiO. As shown in Fig. 6c, the CNF/NiO electrode exhibits high irreversible capacity during the initial cycle, and then the capacity is stabilized on the subsequent cycle. During the initial Li ion charge (insertion) reaction, an obvious plateau voltage for CNF/NiO is observed from 0.9 to 0.3 V. The well-defined voltage plateau in the range of 0.9 V to 0.3 V is due to the main lithiation reaction of CNF/NiO for the conversion reaction to Ni and Li2O and the formation of solid–electrolyte interface (SEI) film. The voltage plateau at around 0.5 V reflects the Li ion charge reaction: NiO + 2Li+ + 2e− ↔ Li2O + Ni. Similar to the CV results, the lithiation plateau moves to a higher voltage of around 1.0 V in the second cycle, which implies that the electrochemical reversibility by the easy polarization after the initial charge cycle. The discharge curves have two slope plateaus at around 1.7 V and 2.2 V, corresponding to the formation of NiO from Ni and Li2O. In the initial cycle, the charge and discharge capacities were 1400 and 950 mA h g−1, respectively. The irreversible capacity of 67.9% in the initial cycle (67.9%) is attributed to the formation of SEI films and on the surfaces of the CNF/NiO, and the intercalation of lithium ions into abundant inner space of woven network interconnected with 3D coaxial CNF/NiO nanofiber. From the second charge–discharge curves, the plateaus are not clear caused by low hysteresis of potential, indicating that the reaction appears to be more reversible. The coulombic efficiency from the second cycle increases steeply to 94.9%, showing that the charge and discharge capacity are 985 and 935 mA h g−1, respectively.
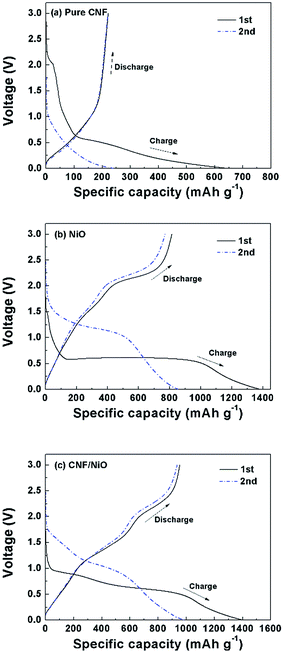 |
| | Fig. 6 Voltage profiles of (a) CNF, (b) NiO, and (c) CNF/NiO at 200 mA g−1 in 1 M LiPF6/EC/DMC. | |
The cyclic voltammograms (CV) of CNF/NiO composite electrodes in the range of 0–3 V at 0.2 mV s−1 scan rate is shown in Fig. 7. For the first scan, the characteristic cathodic peaks at around 0.3 V corresponds to the reduction of NiO to metallic nickel and the formation of reversible SEI layer.33 Two anodic peaks at around 2.4 V and 1.6 V are attributed to the decomposition of Li2O and the electrolyte, respectively.18,34 The variations of main peaks to higher voltage as the cycles continue have a deep relation to the hierarchically porous core–shell structured CNF/NiO with high surface area.34,35
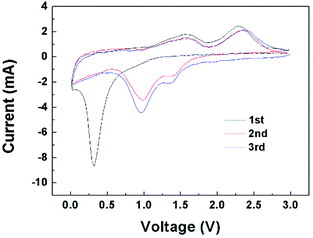 |
| | Fig. 7 Cyclic voltammograms of CNF/NiO at a scanning rate of 0.2 mV s−1 in 1 M LiPF6 EC/DMC electrolyte. | |
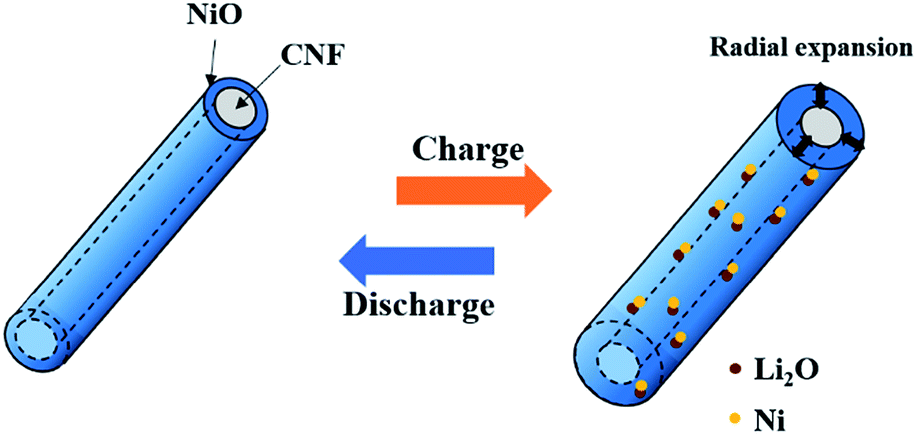
Fig. 8 shows the cycle performance of pure CNF, NiO powder, and CNF/NiO composite. The specific capacity for NiO powder reaches 1370 mA h g−1 in the initial cycle eventually leveling off to 350 mA h g−1 in the 50th cycle due to fatal volume changes, which is near the value of the pure CNF. Compared to the values of pure CNF and NiO powder, the CNF/NiO composites exhibit excellent capacity retention with extremely high values of capacities. A capacity retention of more than 900 mA h g−1 is recorded after the second cycle without an obvious capacity fading except for an initial capacity of 1400 mA h g−1. The specific capacity of CNF/NiO is much higher than the theoretical capacity of 561 mA h g−1 of CNF/NiO. The theoretical capacity of CNF/NiO is calculated as follow: theoretical capacity (TC) of CNF/NiO = TC of NiO × weight% of NiO + TC of graphite × weight% of graphite = 718 × 54.8% + 372 × 45.2% = 561 mA h g−1. The weight% of CNF/NiO obtained from the result of TGA is used in calculating the theoretical capacity of CNF/NiO. The reasons for the high specific capacity and excellent electrochemical stability are as follows. Firstly, the 3D coaxial CNF/NiO connected with the NiO shell on the surface of CNF creates the electrochemical stability. During the charge process, the expansion of the NiO shell is mostly in the radial direction because nanoparticles in the NiO shell are pushed out from the surface of CNF toward the radial direction through inelastic flow (Fig. 9).27,28 The buffering effect by the CNF core enables prohibition of battery failure coming from volume variation by the inelastic NiO shell. Also, the Ni particles, which are converted from the NiO shell through the conversion mechanism (NiO + 2Li+ + 2e− ↔ Li2O + Ni), may be mostly located on the CNF core caused by the facile electron supply of the CNF core. Then the Ni particles can easily transform back into NiO by the CNF core, involving the decomposition of Li2O during the discharge process.36 Thus, the structural stability created by the 3D coaxial structure, achieving good bonding between NiO and CNF, prohibits the fading of capacity resulting from the volume change and pulverization. Secondly, abundant inner space exists in the porous morphology with woven network, which is interconnected with each 3D CNF/NiO coaxial nanocable. The abundant inner space caused by the porous morphology not only offers tremendous channels for the facile electrolyte flow, but also induces excellent contact between the electrolyte and NiO. This characteristic facilitates mass transfer and charge transfer in enhancing the electrochemical rate capability. Thirdly, the 1D CNFs core leads to increased electrical conductivity and mechanical stability. The CNFs play an important role in inducing the potential coupling between the mechanical and electrical networking due to their interconnected morphology between 1D structured carbon fibers. The electrically conductive networking of 1D CNFs facilitates electron transfer, enhancing the electrochemical performance. Fourthly, the CNF core of CNF/NiO gains a lot of pores from the catalytic effect of the NiO shell in the annealing process. The NiO shell consists of NiO nanoparticles with a large grain boundary area. Such pores and grain boundary area provide a larger reaction surfaces and additional intercalation sites for accommodation of lithium ions, leading to higher specific capacity than the theoretical capacity.37–39
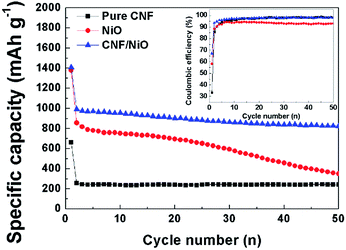 |
| | Fig. 8 Cycle performance of (a) CNF, (b) NiO, and (c) CNF/NiO at 200 mA g−1 in 1 M LiPF6/EC/DMC. | |
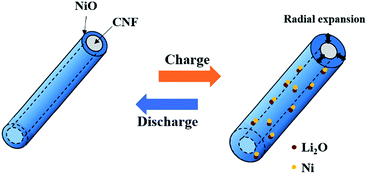 |
| | Fig. 9 Schematic illustration of CNF/NiO during charge–discharge process. | |
Fig. 10 shows the change in the electrochemical impedance spectroscopy (EIS) curve by the Nyquist plots in the range of 100 kHz to 10 mHz for NiO powder, and CNF/NiO electrodes. The internal resistance (RΩ) lies at the intercept of the semicircle in the high frequency region at the real axis. The internal resistances of both NiO powder and CNF/NiO electrodes is almost the same at 2.0 Ω, because there is little difference originating from the intrinsic electrical resistance of the active materials, the electrolyte resistance, and the contact resistance at the interface between the active material and current collector. At the low frequency region, the semicircle is related to charge-transfer resistance (Rct). The charge-transfer resistances of the NiO powder and CNF/NiO electrodes are 55 and 16 Ω, respectively. The charge-transfer resistance of the CNF/NiO electrodes is much smaller than that of NiO powder electrodes, because of (i) its facile lithium ion transfer by abundant inner spaces in NiO shells (ii) increased electrical conductivity by CNFs, and (iii) structural stability by 3D coaxial core–shell morphology. These three factors reduce the ion intercalation distance, facilitate charge transfer, and reduce the resistance. The low charge transfer resistance is beneficial for the enhancement of the electron kinetics in the electrode material and also improves the electrochemical performance of the electrode material.
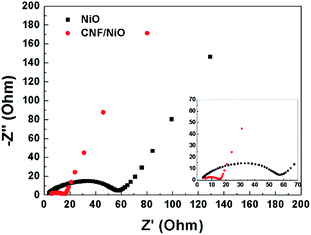 |
| | Fig. 10 AC impedance spectra of NiO and CNF/NiO in 1 M LiPF6/EC/DMC. | |
Fig. 11 shows the capacity retention of the CNF/NiO composite at various current densities. When the current density is 250 mA g−1 at the start, the capacity reaches about 985 mA h g−1. Even with an increased current density of 1000 mA g−1, the CNF/NiO composite electrode delivers a capacity of around 521 mA h g−1. Afterwards, the capacity at 200 mA g−1 delivers 781 mA h g−1 (recovery%: 79.3), indicating good reversibility of the conversion reaction between NiO and Ni. There are three reasons for the excellent retention and good rate capability of CNF/NiO composite. Firstly, a 3D coaxial core–shell CNF/NiO composite facilitates Li insertion and extraction, and ion transfer by offering a smaller resistance and shorter diffusion pathways. Secondly, the improvement of electrical conductivity by CNFs with a 1D pathway promotes a redox reaction. Thirdly, the buffering effect by the elastic CNF core protects radial volume expansion by the inelastic NiO shell.
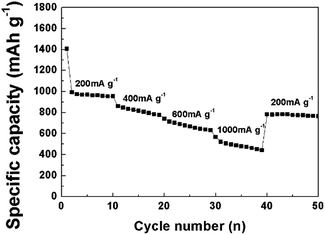 |
| | Fig. 11 Rate performance of CNF/NiO at various current densities from 200 to 1000 mA g−1. | |
3. Experimental
3.1 Preparation of carbon nanofibers
The polymer solution for electrospinning was prepared by dissolving 10 wt% polyacrylonitrile (PAN, Mw = 150000, Aldrich Chemical Co) in N,N-dimethylformamide (DMF), followed by gently stirring for 24 h at 60 °C to obtain a homogeneous solution. The electrospinning process was carried out by a system described in earlier work, which is installed with a power supply (NT-PS-35K, NTSEE, Korea) available for the control of high voltage.40–42 The polymer solution was placed in a 30 ml syringe with a capillary tip of 0.5 mm in inner diameter. The anode of the high voltage power supply was clamped to a syringe needle tip, and the cathode was connected to a metal collector. The electrospun fibers were collected on aluminum foil wrapped around a metal drum rotating at approximately 300 rpm. The applied voltage was 20 kV, the distance between the tip and the collector was 18 cm, and the flow rate of the spinning solution was 1 ml h−1. The electrospun fibers were stabilized by heating to 280 °C at a rate of 1 °C min−1 in air, followed by keeping them at that temperature for 1 h. Finally, the CNFs were prepared by carbonizing the stabilized fibers for 1 h after increasing to 1000 °C at a rate of 5 °C min−1 under nitrogen.
3.2 Preparation of NiO powder
The NiO powder was prepared by calcining 0.5 g of Ni(NO3)2·6H2O for 1 h after increasing to 400 °C at a rate of 5 °C in air, and subsequent cooling down to room temperature. The mechanism of reaction is shown by Ni(NO3)2 → NiO↓ + 2NO2↑ + O2↑.43
3.3 Preparation of coaxial carbon nanofiber/NiO core–shell nanocables
Electrophoretic deposition (EPD) is a reliable method that can easily deposit metal hydroxides such as α-Ni(OH)2 on the surface of a flexible, highly conductive CNF when applying the electric field. In the EPD process, CNF was used as the cathode, and Pt wire as anode. The distance between the two electrodes was 5 cm,f immersed in a Ni(NO3)2·6H2O ethanol solution. After applying a potential of 10 V for 3 h, the sample was washed several times with ethanol, and then dried completely at room temperature. Finally, the CNF/NiO core–shell nanocables were prepared with heat treatment at 300 °C for 2 h under air atmosphere.
3.4 Characterization
The morphologies of NiO powder, pure CNF and CNF/NiO were observed by using a field emission scanning electron microscope (FE-SEM, S-4700, Hitachi, Japan). The particle size and the dispersion degree of NiO on carbon nanofiber were verified using a transmission electron microscope (TEM, JEM-2000 FXII, JEOL, USA) in the Korean Basic Science Institute (KBSI, Gwangju center). The weight loss of CNF/NiO was measured by thermogravimetric analysis (TGA, Shimadzu, TA-50, Japan). The crystallization results for pure CNF, NiO powder and CNF/NiO were analyzed by X-ray diffraction (XRD, D/MAX Ultima III, Rigaku, Japan). Electrochemical charge–discharge behaviors were conducted using coin cells (type CR2032) assembled in an argon-filled glove box. For the fabrication of the working electrodes, a slurry was prepared by mixing the active material (CNF/NiO core–shell nanocables) with super P and poly (acrylic acid) (PAA, Mw = 3000000, Aldrich) at a weight ratio of 70:15:15 in N-methyl pyrrolidinone (NMP). The Super P and PAA were used as conductive additive and binder. The resultant slurry was uniformly pasted on Cu foil with a blade, dried for 12 h at 130 °C in a vacuum oven to completely remove water and pressed between stainless steel twin rollers. Then the foil was punched into circular discs and pressed, and coin cells were assembled with CNF/NiO as the working electrode, Li foil as the counter electrode and a membrane (Celgard 2400) together with glass fiber as a separator. The electrolyte is composed of a solution of 1 M LiPF6 in a mixture of ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:1, v/v) (Techno Semichem Co.). The Li foils were used a reference electrode and a counter one, respectively. Also, the charge–discharge performance of samples was measured by using a two-electrode system. The cyclic voltammetry (CV) was carried out on an IM6e (Jahner Electrik IM6e, Germany) from 0 to 3 V. Electrochemical impedance spectroscopy (EIS) measurements were performed on IM6e (Jahner Electrik IM6e, Germany), and the frequency ranged from 10 mHz to 100 kHz with an applied AC signal amplitude of 5 mV. The charge–discharge test was measured by using a battery cycler system (WBCS 3000, Won-A Tech. Co., Korea).
4. Conclusions
Novel 3D coaxial CNF/NiO core–shell nanocables as anode materials for LIBs were prepared through electrophoretic deposition (EPD) on the surface of CNF and subsequent heat treatment, which is a characteristic of abundant inner spaces in NiO shells on the surface of CNF core. The 3D CNF/NiO coaxial nanocables allows better penetration of electrolyte to enhance the electrochemical performance. The CNF/NiO coaxial nanocable delivers an initial capacity of 1400 mA h g−1 at 200 mA g−1 and maintains a high reversible capacity of 825 mA h g−1 after 50 cycles without showing obvious decay. The reasons for both the excellent retention and good rate capability for CNF/NiO composite is as follows: (i) easy lithium insertion/extraction and facile lithium ion transfer by 3D coaxial core–shell CNF/NiO composite with abundant spaces, (ii) promotion of electron transfer by increased electrical conductivity of 1D, (iii) protection of the radial volume expansion of the inelastic NiO shell by the buffering effect of the elastic CNF core.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (no. 2014R1A2A2A01007540).
References
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novak, Adv. Mater., 1998, 10, 725–763 CrossRef CAS.
- M. Endo, C. Kim, K. Nishimura, T. Fujino and K. Miyashita, Carbon, 2000, 38, 183–197 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- J. Y. Xiang, J. P. Tu, L. Zhang, Y. Zhou, X. L. Wang and S. J. Shi, J. Power Sources, 2010, 195, 313–319 CrossRef CAS PubMed.
- L. L. Xing, C. X. Cui, C. H. Ma and X. Y. Xue, Mater. Lett., 2011, 65, 2104–2106 CrossRef CAS PubMed.
- L. H. Huang, D. Zhu, Y. G. Chen, C. L. Wu, H. B. Sun and H. Yang, Mater. Lett., 2012, 74, 37–39 CrossRef CAS PubMed.
- D. F. Qiu, Z. J. Xu, M. B. Zheng, B. Zhao, L. J. Pan, L. Pu and Y. Shi, J. Solid State Electrochem., 2012, 16, 1889–1892 CrossRef CAS PubMed.
- S. Fan, X. J. Liu, Y. F. Li, E. Y. Yan, C. H. Wang, J. H. Liu and Y. Zhang, Mater. Lett., 2013, 91, 291–293 CrossRef CAS PubMed.
- J. Y. Lee, K. Liang, K. H. An and Y. H. Lee, Synth. Met., 2005, 150, 153–157 CrossRef CAS PubMed.
- J. Park, E. Kang, S. U. Son, H. M. Park, M. K. Lee, J. Kim, K. W. Kim, H. J. Noh, J. H. Park, C. J. Bae, J. G. Park and T. Hyeon, Adv. Mater., 2005, 17, 429–434 CrossRef CAS.
- C. C. Yu, L. X. Zhang, J. L. Shi, J. J. Zhao, J. H. Gao and D. S. Yan, Adv. Funct. Mater., 2008, 18, 1544–1554 CrossRef CAS.
- S. Hosogai and H. Tsutsumi, J. Power Sources, 2009, 194, 1213–1217 CrossRef CAS PubMed.
- H. Pang, Q. Y. Lu, Y. C. Lia and F. Gao, Chem. Commun., 2009, 7542–7544 RSC.
- P. Qin, M. Linder, T. Brinck, G. Boschloo, A. Hagfeldt and L. C. Sun, Adv. Mater., 2009, 21, 2993–2996 CrossRef CAS.
- L. Li, E. A. Gibson, P. Qin, G. Boschloo, M. Gorlov, A. Hagfeldt and L. C. Sun, Adv. Mater., 2010, 22, 1759–1762 CrossRef CAS PubMed.
- A. Debart, L. Dupont, P. Poizot, J. B. Leriche and J. M. Tarascon, J. Electrochem. Soc., 2001, 148, A1266–A1274 CrossRef CAS PubMed.
- J. Fan, T. Wang, C. Z. Yu, B. Tu, Z. Y. Jiang and D. Y. Zhao, Adv. Mater., 2004, 16, 1432–1436 CrossRef CAS.
- L. Yuan, Z. P. Guo, K. Konstantinov, P. Munroe and H. K. Liu, Electrochem. Solid State Lett., 2006, 9, A524–A528 CrossRef CAS PubMed.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacin, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed.
- C. H. Xu, J. Sun and L. A. Gao, J. Power Sources, 2011, 196, 5138–5142 CrossRef CAS PubMed.
- L. Q. Tao, J. T. Zai, K. X. Wang, Y. H. Wan, H. J. Zhang, C. Yu, Y. L. Xiao and X. F. Qian, RSC Adv., 2012, 2, 3410–3415 RSC.
- X. J. Zhu, J. Hu, H. L. Dai, L. Ding and L. Jiang, Electrochim. Acta, 2012, 64, 23–28 CrossRef CAS PubMed.
- J. B. Wu, R. Q. Guo, X. H. Huang and Y. Lin, J. Power Sources, 2014, 248, 115–121 CrossRef CAS PubMed.
- I. Corni, M. P. Ryan and A. R. Boccaccini, J. Eur. Ceram. Soc., 2008, 28, 1353–1367 CrossRef CAS PubMed.
- M. S. Wu, C. Y. Huang and K. H. Lin, Electrochem. Solid State Lett., 2009, 12, A129–A131 CrossRef CAS PubMed.
- S. Santhanagopalan, F. Teng and D. D. Meng, Langmuir, 2011, 27, 561–569 CrossRef CAS PubMed.
- L. Hu, H. Wu, Y. Gao, A. Cao, H. Li, J. McDough, X. Xie, M. Zhou and Y. Cui, Adv. Energy Mater., 2011, 1, 523–527 CrossRef CAS.
- J. W. Wang, X. H. Liu, K. Zhao, A. Palmer, E. Patten, D. Burton, S. X. Mao, Z. Suo and J. Y. Huang, ACS Nano, 2012, 6, 9158–9167 CrossRef CAS PubMed.
- L. Ji, Z. Lin, A. J. Medford and X. Zhang, Chem.–Eur. J., 2009, 15, 10718–10722 CrossRef CAS PubMed.
- L. Ji, Y. Yao, O. Toprakci, Z. Lin, Y. Liang, Q. Shi, A. J. Medford, C. R. Millns and X. Zhang, J. Power Sources, 2010, 195, 2050–2056 CrossRef CAS PubMed.
- B. Wang, J. L. Cheng, Y. P. Wu, D. Wang and D. N. He, Electrochem. Commun., 2012, 23, 5–8 CrossRef CAS PubMed.
- A. K. Rai, L. T. Anh, C. J. Park and J. Kim, Ceram. Int., 2013, 39, 6611–6618 CrossRef PubMed.
- B. Varghese, M. V. Reddy, Z. Yanwu, C. S. Lit, T. C. Hoong, G. V. S. Rao, B. V. R. Chowdari, A. T. S. Wee, C. T. Lim and C. H. Sow, Chem. Mater., 2008, 20, 3360–3367 CrossRef CAS.
- C. Wang, D. L. Wang, Q. M. Wang and H. J. Chen, J. Power Sources, 2010, 195, 7432–7437 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, J. Power Sources, 2001, 97–98, 235–239 CrossRef CAS.
- Y. Wang and Q. Z. Qin, J. Electrochem. Soc., 2002, 149, A873–A878 CrossRef CAS PubMed.
- A. Oya, S. Yoshida, J. Alcaniz-Monge and A. Linares-Solano, Carbon, 1995, 33, 1085–1090 CrossRef CAS.
- Y. Zhu, X. Xiang, E. Liu, Y. Wu, H. Xie, Z. Wu and Y. Tian, Mater. Res. Bull., 2012, 47, 2045–2050 CrossRef CAS PubMed.
- Z. S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 6, 3187–3194 CrossRef PubMed.
- S. H. Park, H. R. Jung, B. K. Kim and W. J. Lee, J. Photochem. Photobiol., A, 2012, 246, 45–49 CrossRef CAS PubMed.
- S. H. Park, H. R. Jung and W. J. Lee, Electrochim. Acta, 2013, 102, 423–428 CrossRef CAS PubMed.
- S. H. Park, B. K. Kim and W. J. Lee, J. Power Sources, 2013, 239, 122–127 CrossRef CAS PubMed.
- T. Battumur, S. B. Ambade, R. B. Ambade, P. Pokharel, D. S. Lee, S. H. Han, W. Lee and S. H. Lee, Curr. Appl. Phys., 2013, 13, 196–204 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.