
Synthesis, characterization and DFT study of oxorhenium(V) complexes incorporating quinoline based tridentate ligands†
Rupa Sarkar,
Amar Hens and
Kajal Krishna Rajak*
Inorganic Chemistry Section, Department of Chemistry, Jadavpur University, Kolkata 700032, India. E-mail: kajalrajak@hotmail.com; kkrajak@chemistry.jdvu.ac.in
First published on 21st January 2015
Abstract
Two tridentate quinoline based Schiff base ligands HL1 and HL2 were prepared by condensation of salicylaldehyde and 2-hydroxy naphthaldehyde with 8-aminoquinoline, respectively in excellent yield. These ligands react with [ReOCl3(OPPh3)(SMe2)] in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in acetone to form mononuclear trans-dichloro oxo complexes of general formula [ReVO(L)Cl2]. Here L− is the deprotonated form of 2-((quinolin-8-ylimino) methyl)phenol (HL1) or 1-[(quinolin-8-ylimino)methyl]naphthalene-2-ol (HL2). The elemental analysis and ESI mass spectroscopic measurements ensure the formation of the desired complexes. The molecular structure of mer-[ReVO(L1)Cl2] was confirmed by single-crystal X-ray diffraction. The complexes were also characterized by different spectroscopic techniques and electrochemical methods. The ground state geometry, NMR and absorption of Re(V) complexes were examined by DFT and TDDFT methods. The natural transition orbital (NTO) and spin density difference map analysis reveal the nature of the excitations. It was also found that the complexes act as a catalyst for oxidation of cis-cyclooctene to the corresponding epoxide by tert-butyl hydroperoxide (TBHP). The NMR and ESI mass spectroscopic analysis ensure the formation of the desired product which was obtained from catalysis.
:1 in acetone to form mononuclear trans-dichloro oxo complexes of general formula [ReVO(L)Cl2]. Here L− is the deprotonated form of 2-((quinolin-8-ylimino) methyl)phenol (HL1) or 1-[(quinolin-8-ylimino)methyl]naphthalene-2-ol (HL2). The elemental analysis and ESI mass spectroscopic measurements ensure the formation of the desired complexes. The molecular structure of mer-[ReVO(L1)Cl2] was confirmed by single-crystal X-ray diffraction. The complexes were also characterized by different spectroscopic techniques and electrochemical methods. The ground state geometry, NMR and absorption of Re(V) complexes were examined by DFT and TDDFT methods. The natural transition orbital (NTO) and spin density difference map analysis reveal the nature of the excitations. It was also found that the complexes act as a catalyst for oxidation of cis-cyclooctene to the corresponding epoxide by tert-butyl hydroperoxide (TBHP). The NMR and ESI mass spectroscopic analysis ensure the formation of the desired product which was obtained from catalysis.
Introduction
The coordination chemistry of high valent rhenium has attracted current attention.1 This is mainly due to the fact that the high-valent oxorhenium complexes are of great importance as oxidation catalysts and because of their capability to transfer an oxygen atom (OAT) to suitable organic substrates2–11 as well as their β-emitting isotopes (186Re and 188Re) used as therapeutic and diagnostic agents in nuclear medicine.12–21The stability and reactivity of such compounds is governed by the close envioronment22,23 about the metal ion and the small change in the ligand architecture can strongly influence its chemistry. The above state of development has prompted us to search new oxorhenium complexes as a part of our general programme on oxorhenium chelates using N, N, O coordinating ligands.24–26
In this paper we describe the syntheses of complexes of types ReVO(NNO)Cl2, using two types of O, N, N-coordinating Schiff base ligands. The structures of the complex, as well as their properties, were probed using X-ray diffraction, spectroscopic and electrochemical techniques. The catalytic property of the complexes was also scrutinized.
In recent years, with the enhancement of the density functional theory (DFT) and especially the improvement of time-dependent density functional theory (TDDFT).27 properties of both ground- and excited-states for medium-sized metal complexes can be calculated at the first-principle level with good accuracy.28,29 In order to get better insight into the geometry and the electronic structure geometry optimizations of the ground was carried out by means of DFT calculations. We also calculate and analyze the singlet state natural transition orbitals (NTOs) derived from TDDFT results and compare them with the ground state molecular orbitals (MOs). The computational modeling of the NMR parameter is also of abiding interest, and such calculation at DFT has emerged as a promising approach for the prediction of nuclear shielding and coupling constants of NMR active nuclei.30 Thus, we have computed the proton and carbon NMR chemical shifts and also the 1H–1H spin–spin coupling constant using the gauge-independent atomic orbital (GIAO)-DFT method, which is aimed at providing the definitive characterization of the complexes.
Experimental section
Materials
[ReOCl3(Me2S)(OPPh3)]31 were prepared as reported in the literature. cis-Cyclooctene and tert-butyl hydroperoxide (tBuOOH) 5.0–6.0 M in n-decane used was purchased from Aldrich Chemical Co. All of the solvents were purified by standard procedures. All the chemicals and solvents were analytically pure and used without further purification. All of the reactions were carried out under a dinitrogen atmosphere.Physical measurements
UV-Vis spectra were recorded on a Perkin-Elmer LAMBDA 25 spectrophotometer. IR spectra were obtained with a Perkin-Elmer L-0100 spectrophotometer. 1H NMR spectra were measured on Bruker FT 300 MHz spectrometer with tetramethylsilane (TMS) as an internal reference. The atom-numbering scheme used for 1H NMR was same as that used in the crystallography. Electrospray ionization mass spectrometry (ESI-MS) spectra were obtained on a Micromass Qtof YA 263 mass spectrometer. Magnetic susceptibilities were measured on a PAR-155 vibrating-sample magnetometer. Elemental analyses (C, H, N) were performed on Perkin-Elmer 2400 series II analyzer and electrochemical measurements were recorded on a CHI 620A electrochemical analyzer using platinum electrode under nitrogen atmosphere. Tetraethylammonium perchlorate (TEAP) was used as a supporting electrolyte and potentials were referenced to the Standard Calomel Electrode (SCE) without junction correction. The cyclic voltammograms were recorded with a scan rate of 50 mV s−1 with iR compensation in all cases.Computational details
The geometrical structures of the ground-state of the selected complexes were optimized by the DFT32 method with B3LYP exchange correlation functional33 approach. The geometry of the complexes was fully optimized in solution phase without any symmetry constraints. There was a good agreement between the theoretical and experimental structures. On the basis of the optimized ground state geometry structure, the absorption property in acetonitrile (CH3CN) media was calculated by time-dependent density functional theory (TDDFT)34 approach associated with the conductor-like polarizable continuum model (CPCM).35 We computed the lowest 40 singlet–singlet transition and results of the TD calculations were qualitatively very similar. The TDDFT approach had been demonstrated to be reliable for calculating spectra properties of many transition metal complexes.36 Due to the presence of electronic correlation in the TDDFT (B3LYP) method it can yield more accurate electronic excitation energies. Hence TDDFT had been shown to provide a reasonable spectral feature for our complex of investigation.In the calculation, the quasirelativistic pseudo potentials of Re atoms proposed by Hay and Wadt37 with 14 valence electrons (outer-core [(5s25p6)] electrons and the (5d6) valence electrons) were employed, and a “double-ξ” quality basis set LANL2DZ was adopted as the basis set for Re atoms. For H we used 6-31(g) basis set and the 6-31+G(d)38 basis set for C, N, O, and Cl atoms for the optimization of the ground state geometries.
In addition, the 1H and 13C NMR properties of the complexes were calculated with the magnetic field perturbation method with the GIAO algorithm39 with the NMR = spin–spin keyword incorporated in the Gaussian 09W program. In calculation, the 6-311+G(2d,p) basis set was employed for all atoms other than rhenium. The relative chemical shift of a given nucleus X in the molecule was defined as δcalcX [ppm] = σcalcX − σrefX where TMS was used as a reference molecule optimized at the same level of theory.39b,40a In order to account for the solvent effect, we used the integral equation-formalism polarizable continuum model (IEFPCM) method.40b,c
Finally to understand the nature of excited states involved in absorption and emission processes natural transition orbital (NTO) analysis had been performed for all complexes. This approach provides the most compact representation of the electronic transitions on terms of an expansion into single particle orbitals by diagonalizing the transition density matrix associated with each excitation.41 The spin density difference map calculations were also performed to explain their optical properties. Figures showing MOs, NTOs and the difference density plots were prepared by using the GaussView 5.1 software. All the calculations were performed with the Gaussian 09W software package.42 GaussSum 2.1 program43 was used to calculate the molecular orbital contributions from groups or atoms.
Crystallographic studies
The single crystal suitable for X-ray crystallographic analysis of the complex [ReVO(L1)Cl2], 1 was obtained by slow evaporation of acetone solution of the complex. The X-ray intensity data were collected on Bruker AXS SMART APEX CCD diffractometer (MoKα, λ = 0.71073 Å) at 293 K. The detector was placed at a distance 6.03 cm from the crystal. Total 606 frames were collected with a scan width of 0.3° in different settings of φ. The data were reduced in SAINTPLUS44 and empirical absorption correction was applied using the SADABS package.44Metal atom was located by Patterson Method and the rest of the non-hydrogen atoms were emerged from successive Fourier synthesis. The structures were refined by full matrix least-square procedure on F2. All non-hydrogen atoms were refined anisotropically. All calculations were performed using the SHELXTL V 6.14 program package.45 Molecular structure plots were drawn using the Oak Ridge thermal ellipsoid plot (ORTEP).46 Relevant crystal data are given in Table 1.
| 1 | |
|---|---|
| a R1 = Σ∣∣Fo∣ − ∣Fc∣∣/Σ∣Fo∣.b wR2 = [Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]]1/2. | |
| Formula | C16H11Cl2N2O2Re |
| Mr | 520.38 |
| Crystal system | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 7.2210 (1) |
| b/Å | 9.5214 (2) |
| c/Å | 12.2908 (2) |
| α/° | 74.277 (1) |
| β/° | 84.604 (1) |
| γ/° | 68.143 (1) |
| V/Å3 | 754.93 (2) |
| Z | 2 |
| Dcalcd/mg m−3 | 2.289 |
| μ/mm−1 | 8.412 |
| θ/° | 1.45–27.53 |
| T/K | 293 (2) |
| Reflns collected | 3456 |
| R1,a wR2b [I > 2σ(I)] | 0.0322, 0.0565 |
| GOF on F2 | 1.000 |
Synthesis of complexes
The complexes [ReVO(L1)Cl2] and [ReVO(L2)Cl2] were prepared by using a general method with [ReOCl3(Me2S) (OPPh3)] as starting materials. Details are given here for a representative case (1 and 2).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) 963, 826, 762, 327, and 307 cm−1. UV-vis [CH3CN; λmax, nm (ε, M−1 cm−1)]: 451 (2740), 358 (4513), 259 (10537), 229 (19373). 1H NMRexptl {300 MHz, DMSO-d6, δ (ppm), J (Hz)}: 8.7 (H15, d, J = 5.1), 8.3 (H7, s), 8.2 (H9, d, J = 7.6), 7.8 (H2, d, J = 8.3) 6.7–8.7 (10H, ArH). 1H NMRtheor {δ (ppm), J (Hz)}: 10.4 (H15, d, J = 6.1), 7.8 (H7, s), 6.7 (H9, d, J = 8.0), 6.9 (H2, d, J = 8.7) 6.5–10.4 (10H, ArH). 13C NMRexptl {500 MHz, CDCl3, δ (ppm)}: 145.45 (CN), 104.7–149.9 (15C, ArC). 13C NMRtheor {δ (ppm)}: 143.6 (CN), 103–149.1 (19C, ArC). Epa (ReVI/ReV couple): 1.44 V (irr), Epc (ligand reduction couple): −0.38 V (irr).O) 962, 832, and 764 cm−1. UV-vis [CH3CN; λmax, nm (ε, M−1 cm−1)]: 456 (9134), 375 (5247), 318 (5452), 256 (18778), 230 (24804). 1H NMRexptl {300 MHz, DMSO-d6, δ (ppm), J (Hz)}: 9.5 (H19, d, J = 5.3), 8.3 (H11, s), 8.2 (H13, d, J = 8.9), 7.6 (H2, d, J = 7.5) 7.6–9.5 (12H, ArH). 1H NMRtheor {δ (ppm), J (Hz)}: 10.5 (H19, d, J = 6.3), 7.8 (H11, s), 7.1 (H13, d, J = 9.7), 6.6 (H2, d, J = 8.0) 6.6–10.5 (12H, ArH). 13C NMRexptl {500 MHz, CDCl3, δ (ppm)}: 143.16 (CN), 105.6–148.3 (19C, ArC). 13C NMRtheor {δ (ppm)}: 143.6 (CN), 103–148.9 (19C, ArC). Epa (ReVI/ReV couple): 1.07 V (irr), Epc (ligand reduction couple): −0.44 V (irr).
O) 963, 826, 762, 327, and 307 cm−1. UV-vis [CH3CN; λmax, nm (ε, M−1 cm−1)]: 451 (2740), 358 (4513), 259 (10537), 229 (19373). 1H NMRexptl {300 MHz, DMSO-d6, δ (ppm), J (Hz)}: 8.7 (H15, d, J = 5.1), 8.3 (H7, s), 8.2 (H9, d, J = 7.6), 7.8 (H2, d, J = 8.3) 6.7–8.7 (10H, ArH). 1H NMRtheor {δ (ppm), J (Hz)}: 10.4 (H15, d, J = 6.1), 7.8 (H7, s), 6.7 (H9, d, J = 8.0), 6.9 (H2, d, J = 8.7) 6.5–10.4 (10H, ArH). 13C NMRexptl {500 MHz, CDCl3, δ (ppm)}: 145.45 (CN), 104.7–149.9 (15C, ArC). 13C NMRtheor {δ (ppm)}: 143.6 (CN), 103–149.1 (19C, ArC). Epa (ReVI/ReV couple): 1.44 V (irr), Epc (ligand reduction couple): −0.38 V (irr).O) 962, 832, and 764 cm−1. UV-vis [CH3CN; λmax, nm (ε, M−1 cm−1)]: 456 (9134), 375 (5247), 318 (5452), 256 (18778), 230 (24804). 1H NMRexptl {300 MHz, DMSO-d6, δ (ppm), J (Hz)}: 9.5 (H19, d, J = 5.3), 8.3 (H11, s), 8.2 (H13, d, J = 8.9), 7.6 (H2, d, J = 7.5) 7.6–9.5 (12H, ArH). 1H NMRtheor {δ (ppm), J (Hz)}: 10.5 (H19, d, J = 6.3), 7.8 (H11, s), 7.1 (H13, d, J = 9.7), 6.6 (H2, d, J = 8.0) 6.6–10.5 (12H, ArH). 13C NMRexptl {500 MHz, CDCl3, δ (ppm)}: 143.16 (CN), 105.6–148.3 (19C, ArC). 13C NMRtheor {δ (ppm)}: 143.6 (CN), 103–148.9 (19C, ArC). Epa (ReVI/ReV couple): 1.07 V (irr), Epc (ligand reduction couple): −0.44 V (irr).Catalytic epoxidation reactions
The catalytic reactions were performed under an atmosphere of nitrogen. The reaction vessel was immersed into an oil bath, and a solution of (1 mmol) of cis-cyclooctene (R) dissolved in 5 ml of CHCl3 and catalyst (10 μmol) were added, and the mixture was heated to 50 °C. During this time the colour of the solution was red. To start the reaction tBuOOH 5.0–6.0 M in n-decane (363 μl, 2 mmol) was added by syringe to the heated solution at once. Suddenly the colour of the solution changes red to colourless. Then the reaction vessel removed from the oil bath as soon as possible. A small amount of manganese dioxide added to destroy excess TBHP, and the mixture was filtered over a small pad of Celite and column was done to obtain the pure product (RO) using 5% ethylacetate in hexane. 1H NMR and ESI mass spectroscopic measurements confirm the epoxidation of cis-cyclooctenethe was well performed by our Re(V) complexes. 1H NMRexptl {300 MHz, CDCl3, δ (ppm)}: 2.3 (H1, m), 2.1 (H2, m), 1.8–1.9 (m, H3, H8), 1.4–1.6 (9H). ESI-MS (CH2Cl2): m/z 127.04 [RO + H]+.Results and discussion
Synthesis
Two tridentate ligands HL1 and HL2 (general abbreviation, HL) used in this work are given in Chart 1 where L1 and L2 are the deprotonated form of the ligands. The two Schiff base ligands [Chart 1] were prepared according to the literature procedure47,48 The ligands were used as a monoanionic N, N, O coordinating agents for the preparation of the Re(V) complexes (Scheme 1). | ||
| Chart 1 | ||
 | ||
| Scheme 1 Schematic representation for the synthesis of the complexes. | ||
The stoichiometric reaction of [ReOCl3(Me2S)(OPPh3)] with HL1 and HL2 in a ratio of 1:1 in acetone afforded the brown coloured complex, [ReVO(L1)Cl2], and red colored species of formula [ReVO(L2)Cl2], respectively in excellent yields (Scheme 1). The elemental analysis and ESI mass spectroscopic measurements confirm the formation of the synthesized complexes. (see Experimental section).
Crystal structure
The molecular structures of complexes [ReVO(L1)Cl2], 1 was determined by single-crystal X-ray diffractometry. The complex crystallizes in P space group. The selected bond distances and angles for 1 are listed in Table 2, and the molecular views are shown in Fig. 1.
| Bond lengths (Å) | |||||
|---|---|---|---|---|---|
| Re1–N1 | 2.177(4) | Re1–O2 | 1.934(3) | C7–N1 | 1.318(5) |
| Re1–N2 | 2.123(4) | Re1–Cl1 | 2.3853(13) | ||
| Re1–O1 | 1.675(3) | Re1–Cl2 | 2.3653(14) | ||
|
|||||
| Bond angles (°) | |||||
| Cl1–Re1–Cl2 | 169.44(5) | N1–Re1–Cl2 | 87.91(10) | N2–Re1–Cl2 | 90.19(11) |
| O1–Re1–O2 | 113.07(15) | N1–Re1–N2 | 74.47(14) | O1–Re1–Cl1 | 93.45(13) |
| N1–Re1–O1 | 162.08(15) | N2–Re1–O1 | 88.07(16) | O1–Re1–Cl2 | 96.27(13) |
| N1–Re1–O2 | 84.58(13) | N2–Re1–O2 | 158.73(14) | O2–Re1–Cl1 | 87.03(11) |
| N1–Re1–Cl1 | 84.08(10) | N2–Re1–Cl1 | 94.27(11) | O2–Re1–Cl2 | 85.03(11) |
 | ||
| Fig. 1 ORTEP plot and atom labeling scheme of 1. Hydrogen atoms are omitted for clarity. | ||
In the complex 1, the ligand (HL1) behaves as O, N, N coordinating monoanionic ligand. The geometry around the Re(V) metal center is distorted octahedral and this is characterized by Cl1–Re1–Cl2 bond angle of 169.44(5) for 1. The ligand coordinates in a tridentate meridional mode, while the oxo groups are trans to the imino nitrogen (N1) atoms completing the basal plane. The chlorine atoms occupy the apical positions of the octahedron and are thus trans to each other. A limited number of trans dichloro oxorhenium(V) with a tridentate N2O coordinating ligand is reported in the literature.49
It is believed that the meridional binding mode.50 of three chlorine atoms in the starting material [ReOCl3(OPPh3)(SMe2)]31 helps to achieve the rare ReV trans-dichloro oxo complexes. In the complex 1, N2–Re1–O2 angle being 158.73(14), significantly deviated from the value of 180°. The rhenium oxygen bond Re1–O1 was observed at 1.675(3), confirming a triple-bond character.51 The Re1–O2 is found at 1.934(3). The pyridyl N2–Re1 bond is shorter than the imino N1–Re1 bond, which is in agreement with the trans influence induced by O1. Both apical chloro ligands display bond distances to the central metal similar to those reported in the literature.52–55a
IR spectra
The IR spectra of the complexes were recorded in a KBr disk. The characteristic IR data were given in the Experimental section. Both the complexes show one ReO stretching frequency in the range 962–963 cm−1. The CN stretching frequency were observed 1606 cm−1 and 1592 cm−1 in complex 1 and 2 respectively.
NMR spectra
The complexes are diamagnetic corresponding to the 5dxy2 configuration and display well resolved 1H and 13C NMR spectra in DMSO-d6 solution.55b,c The NMR spectra were assigned by considering chemical shift values, spin–spin intensity, and spin–spin-coupling patterns. The signals of the protons close to the O, N, N coordinating atoms are all shifted downfield with respect to those of the free ligands.H15 (for complex 1) and H19 (for complex 2) are observed as a doublet in the range 8.7–9.5 ppm. The azo methine proton H7 (for complex 1) and H11 (for complex 2) are observed as a singlet at 8.3 ppm. For complex 1, H9 is observed at ∼6.7 ppm as a doublet. Whereas H13 is observed at ∼7.6 ppm as a doublet in case of complex 2. All the aromatic protons span in the range ∼6.7–8.7 ppm in complex 1, whereas, in complex 2 all the aromatic protons span in the range ∼7.6–9.5 ppm.
The experimentally observed values are well-correlated with calculated values. The correlation diagram between the calculated and experimental 1H NMR chemical shift is given in ESI (Fig. S1†) and nJH,H coupling constants for complex 1 and 2 is shown in Fig. 2(a). 13C NMR spectra of the complexes were also recorded, and the assigned peaks are given in the Experimental section. The imine carbon was observed near about 143 ppm and the aromatic carbon span in the region 150–103 ppm. The calculated NMR spectral chemical shifts agree well with the experimental data. The experimental and calculated NMR data are given in the Experimental section. The correlation between the experimental and calculated 13C NMR chemical shift of 2 is shown in Fig. 2(b) as a representative case.
 | ||
Fig. 2 (a) Linear correlation between the experimental and calculated nJ(H,H) of 1 ( ) and 2 ( ) and 2 ( ). (b) Linear correlation between the experimental and calculated 13C NMR chemical shifts of 2. ). (b) Linear correlation between the experimental and calculated 13C NMR chemical shifts of 2. | ||
Geometry optimization, electronic structure
The complexes are diamagnetic; thus the geometry optimizations of two complexes were done in their singlet spin state. The molecular structures of [ReVO(L1)Cl2] and [ReVO(L2)Cl2] have been fully optimized in their singlet spin states. Main optimized geometrical parameters of 1 and 2 are given in Table 3. The optimized structures of the complexes 1 and 2 at singlet state are shown in Fig. 3. The modeled geometries possess a distorted octahedral arrangement around the Re(V) center. The geometry used for the ground state optimization is based on crystal structure parameter for complex 1, but we are unable to grow the single crystal suitable for X-ray structure determination in case of complex 2. Therefore the significant bond distances and angles of the optimized geometry of the complexes in solution phase are compared with the crystal structure parameter of 1.| Bond lengths (Å) | |||
|---|---|---|---|
| 1 | 2 | ||
| Re1–N1 | 2.189 | Re1–N1 | 2.164 |
| Re1–N2 | 2.124 | Re1–N2 | 2.121 |
| Re1–O1 | 1.718 | Re1–O1 | 1.721 |
| Re1–O2 | 1.942 | Re1–O2 | 1.939 |
| Re1–Cl1 | 2.445 | Re1–Cl1 | 2.457 |
| Re1–Cl2 | 2.451 | Re1–Cl2 | 2.447 |
| C7–N1 | 1.328 | C11–N1 | 1.338 |
|
|||
| Bond angles (°) | |||
| 1 | 2 | ||
| Cl1–Re1–Cl2 | 169.66 | Cl1–Re1–Cl2 | 170.56 |
| O1–Re1–O2 | 115.36 | O1–Re1–O2 | 116.21 |
| N1–Re1–O1 | 161.78 | N1–Re1–O1 | 162.02 |
| N1–Re1–O2 | 82.78 | N1–Re1–O2 | 81.75 |
| N1–Re1–Cl1 | 84.35 | N1–Re1–Cl1 | 86.86 |
| N1–Re1–Cl2 | 86.84 | N1–Re1–Cl2 | 85.28 |
| N1–Re1–N2 | 75.35 | N1–Re1–N2 | 75.62 |
| N2–Re1–O1 | 86.57 | N2–Re1–O1 | 86.44 |
| N2–Re1–O2 | 157.96 | N2–Re1–O2 | 157.28 |
| N2–Re1–Cl1 | 93.47 | N2–Re1–Cl1 | 89.87 |
| N2–Re1–Cl2 | 89.51 | N2–Re1–Cl2 | 93.23 |
| O1–Re1–Cl1 | 94.70 | O1–Re1–Cl1 | 94.45 |
| O1–Re1–Cl2 | 95.34 | O1–Re1–Cl2 | 94.62 |
| O2–Re1–Cl1 | 87.02 | O2–Re1–Cl1 | 86.91 |
| O2–Re1–Cl2 | 86.50 | O2–Re1–Cl2 | 86.78 |
 | ||
| Fig. 3 Optimized molecular structures of 1 and 2 at ground state. (Re: Cyan, N: Blue, O: Red, C: Grey, H: White.). | ||
As depicted in Fig. 3, there is distorted octahedral coordination geometry around the rhenium center. The crystal structure data of 1 (Table 2) is in good agreement with optimized parameters and the slight discrepancy (maximum deviation 0.08 Å for Re1–Cl2 bond distance) comes from the crystal lattice distortion existing in real molecules. The optimized geometries of the complexes do not show significant differences in the coordinate sphere around the metal center. It reveals that the ligands bind in a similar fashion in the complexes.
The isodensity plot from HOMO−3 to LUMO+3 for 1 is shown in Fig. 4 (same plot for 2 is given in ESI (Fig. S2†)). The partial frontier molecular orbital compositions and energy levels of 1 are listed in Table 4 while that for 2 is given in ESI (Table S1†).
 | ||
| Fig. 4 Isodensity plots of frontier orbitals of [ReVO(L1)Cl2]. | ||
| Orbital | Energy (eV) | Contribution (%) | Main bond type | ||||
|---|---|---|---|---|---|---|---|
| Re | Cl | O |
Aromatic system | Imine | |||
| L+5 | −1.3967 | 1 | 1 | 0 | 97 | 1 | π*(L1) |
| L+4 | −1.4294 | 53 | 32 | 4 | 8 | 3 | d(Re) + p(Cl) |
| L+3 | −2.2019 | 6 | 0 | 0 | 75 | 18 | π*(L1) |
| L+2 | −2.9459 | 53 | 1 | 18 | 25 | 3 | d(Re) + π*(O) + π*(L1) |
| L+1 | −3.0974 | 17 | 1 | 6 | 57 | 18 | d(Re) + π*(L1) |
| LUMO | −3.5358 | 52 | 7 | 22 | 13 | 7 | d(Re) + π*(O) + π*(L1) |
| HOMO | −6.0698 | 50 | 18 | 1 | 28 | 4 | d(Re) + p(Cl) + π(L1) |
| H−1 | −6.6777 | 2 | 6 | 1 | 80 | 10 | π(L1) |
| H−2 | −7.2429 | 6 | 22 | 0 | 69 | 3 | p(Cl) + π(L1) |
| H−3 | −7.9158 | 0 | 9 | 1 | 90 | 1 | π(L1) |
| H−4 | −7.9694 | 0 | 47 | 4 | 45 | 4 | p(Cl) + π(L1) |
| H−5 | −8.0802 | 0 | 41 | 7 | 46 | 6 | p(Cl) + π(L1) |
The HOMO (H) and H−1 are lying 0.60 and 0.73 eV for complex 1 and complex 2, respectively. In both the complex, the electron density in HOMO mainly reside on aromatic moiety (in the range 28–53%) and metal d orbital (in the range 35–50%) along with some chlorine p orbital (10–18%). In case of complex 1, the electron density mainly in H−1 reside on ligand moiety (80%) while for complex 2, H−1 mainly composed of ligand moiety (59%) and metal d orbital (22%) and some chlorine p orbital (17%). In case of 2 the HOMO is less stabilized by an amount ∼1.52 eV compared to 1. For complex 1 the energy difference between HOMO and LUMO (L) occurs at 2.53 eV while for complex 2 the same occurs at 2.29 eV and this energy gap is minimum for 2 and maximum for 1. In complex 1 and complex 2 the H−2 is lying ∼1.17 eV and 0.88 eV, respectively below the HOMO. For complex 1, H−2 is mainly composed of ∼69% ligand π orbital and 22% of p(Cl) orbital while for complex 2 predominant contribution arises from ligand π orbital. The HOMO−3 and HOMO−4 are almost degenerate (energy difference ∼0.05 eV in 1 and ∼0.31 eV in 2). LUMO+1 and LUMO+2 are lying in the range 0.43–0.44 and 0.58–0.72 eV, respectively, above the LUMO for the complexes. The electron densities of LUMO, LUMO+1, and LUMO+2 are mainly distributed between the metal d orbitals and the ligand frame. LUMO+3 is completely delocalized over the ligand moiety.
Absorption spectral properties
The absorption spectra of the complexes were recorded in acetonitrile (CH3CN) solution at room temperature and display four well resolved peaks for 1. Complex 2 displays five well resolved peaks. For complex 1 these are observed at 451, 358, 259 and 229 nm while that for complex 2 occurs at 456, 375, 318, 256 and 230 nm. The band within the range 375–456 nm can reasonably assigned to the classical d–d transition and ILCT transition. The absorption band in the region 318–358 nm are believed to be of LMCT in origin. The band near 259 nm (for complex 1) and 256 nm (for complex 2) are mainly ILCT character along with a little MLCT character. The highest energy bands with maxima in the region 229–230 nm for complexes are mainly admixture of ILCT character along with a little LMCT character. These assignments were supported by theoretical calculations (from spin density plot and also from NTO analysis). The position of bands and the corresponding Δε values are given in the Experimental section.Fig. 5 illustrates the experimental spectrum for complex 1 and 2. The calculated absorption energies associated with their oscillator strengths, the main configurations and their assignments as well as the experimental result of 1 is given in Table 5 while that for 2 is given in ESI (Table S2†).
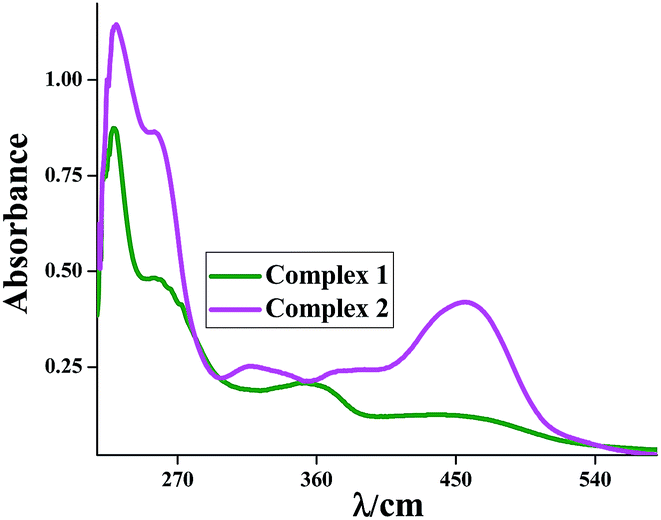 | ||
| Fig. 5 Experimental absorption spectra of 1 and 2 in acetonitrile solution. | ||
| Excitation | Composition | E (eV) | Oscillator strength (f) | λtheo (nm) | Assign | λexp (nm) |
|---|---|---|---|---|---|---|
| 1 | HOMO−1 → LUMO (78%) | 2.4855 | 0.0892 | 498.84 | LMCT/ILCT/LLCT | 451 |
| HOMO−1 → LUMO+1 (5%) | LMCT/ILCT/LLCT | |||||
| HOMO → LUMO+1 (10%) | d–d/ILCT/LLCT | |||||
| HOMO → LUMO+2 (3%) | d–d/ILCT/LLCT | |||||
| 2 | HOMO−2 → LUMO (3%) | 2.8450 | 0.0427 | 435.79 | LMCT/ILCT/LLCT | |
| HOMO−1 → LUMO (2%) | LMCT/ILCT/LLCT | |||||
| HOMO−1 → LUMO+1 (28%) | LMCT/ILCT/LLCT | |||||
| HOMO−1 → LUMO+2 (60%) | LMCT/ILCT/LLCT | |||||
| 3 | HOMO−7 → LUMO (4%) | 3.5886 | 0.0154 | 345.49 | LMCT/ILCT/LLCT | 358 |
| HOMO−6 → LUMO (8%) | LMCT/ILCT/LLCT | |||||
| HOMO−5 → LUMO (7%) | LMCT/ILCT/LLCT | |||||
| HOMO−4 → LUMO (57%) | LMCT/ILCT/LLCT | |||||
| HOMO−3 → LUMO (11%) | LMCT/ILCT/LLCT | |||||
| HOMO−2 → LUMO+1 (3%) | LMCT/ILCT/LLCT | |||||
| HOMO−2 → LUMO+2 (2%) | LMCT/ILCT/LLCT | |||||
| 4 | HOMO−9 → LUMO+1 (25%) | 4.8163 | 0.0109 | 257.43 | LMCT/ILCT/LLCT | 259 |
| HOMO−9 → LUMO+2 (58%) | LMCT/ILCT/LLCT | |||||
| HOMO−8 → LUMO+1 (4%) | LMCT/ILCT/LLCT | |||||
| HOMO → LUMO+6 (5%) | MLCT/ILCT/LLCT | |||||
| 5 | HOMO−12 → LUMO (52%) | 4.9805 | 0.0022 | 248.94 | LMCT/ILCT/LLCT | 229 |
| HOMO−12 → LUMO+1 (3%) | LMCT/ILCT/LLCT | |||||
| HOMO−11 → LUMO (4%) | LMCT/ILCT/LLCT | |||||
| HOMO−10 → LUMO+1 (29%) | LMCT/ILCT/LLCT | |||||
| HOMO → LUMO+7 (2%) | ILCT/LLCT |
Fig. 6 shows the accompanying electron density redistributions in complex 1 and complex 2. From spin density plot we can conclude that the absorption bands in the region 375–456 nm have mixed d–d and ILCT character.
 | ||
| Fig. 6 Difference electron density upon excitation from the ground state (S0) to allowed singlet states (50 singlet to singlet excitations) for complexes 1 and 2 determined with TD-DFT (B3LYP/CPCM-CH3CN) calculations. Turquoise and purple colors show regions of decreasing and increasing electron density, respectively. | ||
In order to analyze the nature of absorption, we performed an NTO analysis based on the calculated transition density matrices.41 This method offers the most compact representation of the transition density between the ground and excited states in terms of an expansion into single-particle transitions (hole and electron states for each given excitation).
Here we refer to the unoccupied and occupied NTOs as “electron” and “hole” transition orbitals, respectively. Note that NTOs are not the same as virtual and occupied MO pairs from the ground state calculations. Fig. 7 illustrates the natural transition orbitals (NTOs) for complex 1 while the same for complex 2 is given in ESI (Fig. S3†).
 | ||
| Fig. 7 Natural transition orbitals (NTOs) for the complex 1 illustrating the nature of optically active singlet excited states in the absorption bands 451, 358, 259 and 229 nm. For each state, the respective number of the state, transition energy (eV), and the oscillator strength (in parentheses) are listed. Shown are only occupied (holes) and unoccupied (electrons) NTO pairs that contribute more than 25% to each excited state. | ||
Based on our TDDFT NTOs analysis the bands in the lowest energy region 451 nm (for complex 1) and 375–456 (for complex 2) can be characterized as an admixture of classical d–d transition and ILCT transition. As illustrated in Fig. 7, optical excitations occur from the occupied (hole) transition orbitals to the unoccupied (electron) transition orbitals. In case of lowest energy bands for both the complexes the hole NTOs are localized on the Re center along with π orbitals of ligands (t2g–π) while the electron NTOs are also delocalized over the Re center and π*orbital of the ligand moiety (t2g–π*).
Electrochemical studies
Cyclic voltammetry was performed for both the complexes in dry acetonitrile solution at room temperature under nitrogen atmosphere with tetraethylammonium perchlorate (TEAP) as the supporting electrolyte using a Pt electrode as working electrode and the potentials are referenced to the saturated calomel electrode (SCE) without junction correction. The representative spectra of complex 1 and 2 are given in ESI (Fig. S4 and S5†).Both the complexes exhibit a single ligand centered reduction wave and a metal centered oxidation wave (ReVI/ReV), both of which are electrochemically irreversible in nature. Both the complexes exhibited irreversible oxidations wave at ∼1.44 V for 1 and ∼1.07 V for 2, which can be assigned to one-electron oxidations ReV → ReVI. The oxidation potential of complex 2 is lesser than the oxidation potential of complex 1. This result arises may be due to the presence of naphthalene ring in case of complex 2. Due to presence of naphthalene ring electron density at metal increases, rendering the complex 2 more easy to oxidize. It is also concluded from here the cathodic shift in case of complex 2 compared to the complex 1 can be attributed to the destabilization of the HOMO of the complex 2. Both the complexes show irreversible reductions wave at ∼−0.39 V for 1 and ∼−0.44 V for 2 where the L2 is more difficult to reduce.
Catalytic epoxidations
Our interest in epoxidation reactions employing Re(V) compounds56–58 led us to explore compounds 1 and 2 as catalysts in the epoxidation of cis-cyclooctene with tert-butyl hydroperoxide (TBHP) as the oxidant. Rhenium(V) compounds are potentially ideal catalysts as they are conveniently prepared and are easy to handle in ambient conditions. In particular, the latter would be advantageous over Re(VII) systems that tend to be more difficult to handle.Typically, the catalyst (1 equiv.) and cis-cyclooctene (R) (100 equiv.) were mixed in chloroform and heated to 50 °C, whereupon the catalyst slowly dissolved. The oxidant (TBHP in 5.0–6.0 M in n-decane, 200 equiv.) was then added, which initiated the reaction. After addition of the peroxide a color change from red to colourless was apparent. A small amount of manganese dioxide added to destroy excess TBHP, and the mixture was filtered over a small pad of Celite and column chromatography was done to obtain the pure product using 5% ethylacetate in hexane. 1H NMR and ESI mass spectroscopic measurements confirm the epoxidation of cis-cyclooctene (Scheme 2) was well performed by our Re(V) complexes. The analysis of this product (RO) by 1H NMR and ESI mass spectra revealed a clean catalytic reaction. In particular, 2-tert-butoxycyclooctanol is absent, a possible side product resulting from ring opening of the epoxide by nucleophilic attack of the formed tert-butanol. However, the catalytic performance of compounds is not comparable to the that of, for example, MTO.59,60 Whether the mechanism involves a Re(VII) or a Re(V) species is unclear because we were not able to isolate any pure rhenium compounds after the epoxidation reactions. Herrmann and co-workers have shown that the catalytically active species in epoxidations with MTO involves the 7-coordinate bisperoxorhenium(VII) complex [ReO2(O2)2(H2O)].61 In contrast, the catalytically active species in the few epoxidations involving other than rhenium(VII) compounds is less clear, but oxidation to a rhenium(VII) peroxo species is suggested.62,63 Thus, for the formation of the active catalyst, the complex 1 and complex 2 may be oxidized to the [ReO3 (L1)] and [ReO3 (L2)], respectively.
 | ||
| Scheme 2 Catalytic epoxidation of cyclooctene. Conditions: 1 mol% Re(V) catalyst 1–2, 1 equiv. of cyclooctene (R), 2 equiv. of tBuOOH (5.5 M in decane), 50 °C, CHCl3. | ||
The 1H NMR data and ESI mass values are given in the Experimental section. The 1H NMR and ESI mass spectrum of the product which was obtained by the catalytic reaction are given in the ESI (Fig. S6 and S7†).
From mass and 1H NMR spectral studies it reveals that the epoxidation of cis-cyclooctene was take place. To get a better understanding, here we also performed UV-vis study to establish the mechanistic path. In UV-vis study we can easily measure the formation of catalytic active species. Both of the complexes showed similar pattern in UV-vis study of the catalytic process; here compound 2 was taken as a representative case. The whole process was carried out using chloroform as a solvent. The stock solution was prepared with the help of 0.2 μmol of compound 2 (1 equiv.), 20 μmol of cis-cyclooctene (100 equiv.), and 2 g of chloroform were mixed in a quartz cuvette, which was placed into a spectrometer equipped with a holder thermostat controlled at 50 °C. In order to increase the rate of the diluted reaction, excess oxidation reagent was added (800 μmol of TBHP, 3000 equiv.). Immediately after addition of TBHP the first UV-vis spectrum was measured, and thereafter, every 10 min additional spectra were acquired for up to 5 h. Fig. 8 shows UV-vis spectra between 300 and 550 nm. The spectrum (black color) shows a strong absorption at 455 nm was representative spectra of isolated compound 2. In addition with cis-cyclooctene and TBHP with the catalyst drastic changes in UV-vis spectra was observed. The absorption intensity at 455 nm undergoes some blue shift at 424 nm and a new band appeared at 365 nm. We carried out time dependent UV-vis spectra on this mixture with 10 min interval of time and it was observed that both of the absorption intensity of the new peaks undergoes decreases with time. As shown in inset of Fig. 8(a) depicted that the rate of decreasing of absorption intensity was faster in initial part of the investigation and becomes saturated near at 80 min which implies that the ending point of the reaction. These result suggested that some intermediate catalytic species was generated.
 | ||
| Fig. 8 Catalytic epoxidation of cyclooctene, 1 mol% catalyst 2, 1 equiv. of cyclooctene, 30 equiv. of TBHP; (a) UV-vis spectra measured every 10 min. The insert shows the absorbance at 365 nm. | ||
A literature survey was carried out to determine the nature of the formed intermediate catalytic compound. It seems reasonable to assign the UV-vis absorption at 365 nm which may be arise due to formation of the catalytic species rhenium trans-dioxo compound, as comparable complexes absorb in the same wavelength range ([Re(O)2(py)4]+) λmax = 331 nm,64 [Re(O)2(4-MeOPy)4]+ and [Re(O)2(4-pyrrolidinopyridine)4]+ λmax = 334 nm,65 [Re(O)2(acyclic tetraamine)]+ λmax = 350 nm,66 [Re(O)2(tridentate O2 ligand)] λmax = 300–350 nm.67
Conclusion
In summary, we have synthesized monomeric Re(V) complexes with two different tridentate Schiff base ligands. The complexes are characterized by different spectroscopic techniques, elemental analysis and X-ray structure determination. The present work investigated the ground state geometry, NMR, and absorption properties of two Re(V) complexes by DFT and TDDFT methods. From our calculation results, we have characterized all of the low-lying electronic states as admixture of ILCT, MLCT and LLCT in character. The nature of the transitions was also supported by Spin density difference map and natural transition orbital (NTO) analysis. Catalytic property of both the complexes was also scrutinized.Acknowledgements
Financial assistance received from the Council of Scientific and Industrial Research, New Delhi, India, Science & Engineering Research Board, New Delhi, India is gratefully acknowledged. We are also thankful to Department of Science and Technology, New Delhi, India for the data collection on the CCD facility setup (Jadvpur University) under DST-FIST program. Rupa Sarkar and Amar Hens are also thankful to UGC, New Delhi for the research fellowship.References
- (a) J. Bernard, K. Ortner, B. Spingler, H. J. Pietzsch and R. Alberto, Inorg. Chem., 2003, 42, 1014 CrossRef CAS PubMed; (b) H. J. Lee and W. M. Partridge, Bioconjugate Chem., 2003, 14, 546 CrossRef CAS PubMed; (c) J. B. Arterburn, K. V. Rao, D. M. Goreham, M. V. Valenzuela, M. S. Holguin, K. A Hall, K. C. Ott and J. C. Bryan, Organometallics, 2000, 19, 1789 CrossRef CAS; (d) R. Sarkar, P. Mondal and K. K. Rajak, Dalton Trans., 2014, 43, 2859 RSC.
- (a) S. S. Jurisson and J. D. Lydon, Chem. Rev., 1999, 99, 2205 CrossRef CAS PubMed; (b) W. A. Volkert and T. J. Hoffman, Chem. Rev., 1999, 99, 2269 CrossRef CAS PubMed; (c) J. R. Dilworth and S. J. Parrott, Chem. Soc. Rev., 1998, 27, 43 RSC.
- (a) E. John, M. L. Thakur, J. De Fulvio, M. R. McDevitt and I. Dajanov, J. Nucl. Med., 1993, 34, 260 CAS; (b) J. P. DiZio, C. J. Anderson, A. Davison, G. J. Ehrhardt, K. E. Carlson, M. J. Welch and J. A. Katzenellenbogen, J. Nucl. Med., 1992, 33, 558 CAS.
- (a) G. L. Griffiths, D. M. Goldenberg, A. Jones and H. Hansen, J. BioConjugate Chem., 1992, 3, 91 CrossRef CAS; (b) M. J. Clarke and L. Podbielsky, Coord. Chem. Rev., 1987, 78, 253 CrossRef CAS.
- (a) Technetium and Rhenium in Nuclear Medicine, ed. M. Nicolini, G. Bandoli and U. Mazzi, S. G. Editoriali, Padova, Italy, 1995, vol. 4 Search PubMed; (b) K. Libson, L. Helm, A. Roodt, C. Cutler, A. E. Merbach, J. Sullivan and E. Deutch, in Technetium and Rhenium in Chemistry and Nuclear Medicine, ed. M. Nicolini, G. Bandoli and U. Mazzi, Raven Press, New York, 1989, vol. 3 Search PubMed.
- (a) M. Papachristou, I. C. Pirmettis, C. Tsoukalas, D. Papagiannopoulou, C. Raptopoulou, A. Terzis, C. I. Stassinopoulou, E. Chiotellis, M. Pelecanou and M. Papadopoulos, Inorg. Chem., 2003, 42, 5778 CrossRef CAS PubMed; (b) L. Hansen, S. Hirota, X. Xu, A. T. Taylor and L. G. Marzilli, Inorg. Chem., 2000, 39, 5731 CrossRef CAS.
- (a) A. R. Cowley, J. R. Dilworth and P. S. Donnelly, Inorg. Chem., 2003, 42, 929 CrossRef CAS PubMed; (b) A. R. Cowley, J. R. Dilworth, P. S. Donnelly and J. Woollard Shore, J. Chem. Soc., Dalton Trans., 2003, 748 RSC; (c) R. Visentin, R. Rossin, M. C. Giron, A. Dolmella, G. Bandoli and U. Mazzi, Inorg. Chem., 2003, 42, 950 CrossRef CAS PubMed.
- (a) C. Rader and B. List, Chem.–Eur. J., 2000, 6, 2091 CrossRef CAS; (b) C. J. Anderson and M. J. Welch, Chem. Rev., 1999, 99, 2219 CrossRef CAS PubMed; (c) D. E. Reichert, J. S. Lewis and C. J. Anderson, Coord. Chem. Rev., 1999, 184, 3 CrossRef CAS; (d) P. Traar, J. A. Schachner, L. Steiner, A. Sachse, M. Volpe and N. C. Mösch-Zanetti, Inorg. Chem., 2011, 50, 1983–1990 CrossRef CAS PubMed.
- (a) L. G. Marzilli, M. G. Banaszczyk, L. Hansen, Z. Kuklenyik, R. Cini and A. Taylor Jr, Inorg. Chem., 1994, 33, 4850 CrossRef CAS; (b) L. Hansen, Y. D. Lampeka, S. P. Gavrish, X. Xu, A. T. Taylor and L. G. Marzilli, Inorg. Chem., 2000, 39, 5859 CrossRef CAS; (c) B. Machura, M. Wolff, E. Benoist, J. A. Schachnerd and N. C. Mösch-Zanettid, Dalton Trans., 2013, 42, 8827 RSC; (d) J. L. Smeltz, C. P. Lilly, P. D. Boyle and E. A. Ison, J. Am. Chem. Soc., 2013, 135, 9433–9441 CrossRef CAS PubMed.
- (a) C. Tessier, F. D. Rochon and A. L. Beauchamp, Inorg. Chem., 2002, 41, 6527 CrossRef CAS PubMed; (b) S. Fortin and A. L. Beauchamp, Inorg. Chem., 2000, 39, 4886 CrossRef CAS; (c) M. V. Bereau, S. I. Khan and M. M. Abu-Omar, Inorg. Chem., 2001, 40, 6767 CrossRef PubMed.
- D. Parker, in Comprehensive Supramolecular Chemistry, ed. D. N. Reinhoudt, Pergamon Press, Cambridge, 1996, vol. 10 Search PubMed.
- P. D. Benny, C. L. Barnes, P. M. Piekarski, J. D. Lydon and S. S. Jurisson, Inorg. Chem., 2003, 42, 6519 CrossRef CAS PubMed.
- (a) K. J. C. van Bommel, W. Verboom, R. Hulst, H. Kooijman, A. L. spek and D. N. Reinhoudt, Inorg. Chem., 2000, 39, 4099 CrossRef CAS; (b) K. J. C. van Bommel, W. Verboom, H. Kooijman, A. L. Spek and D. N. Reinhoudt, Inorg. Chem., 1998, 37, 4197 CrossRef CAS PubMed.
- (a) W. A. Herrmann, M. U. Rauch and G. R. J. Artus, Inorg. Chem., 1996, 35, 1988 CrossRef CAS; (b) G. Laurenczy, F. Lukacs, R. Roulet, W. A. Herrmann and R. W. Fischer, Organometallics, 1996, 15, 848 CrossRef CAS.
- (a) F. Tisato, F. Refosco, U. Mazzi, G. Bandoli and M. Nicolini, Inorg. Chim. Acta, 1991, 189, 97 CrossRef CAS; (b) F. Tisato, F. Refosco, U. Mazzi, G. Bandoli and A. Dolmella, Inorg. Chim. Acta, 1989, 164, 127 CrossRef CAS.
- (a) J. M. Botha, K. Umakoshi and Y. Sasaki, Inorg. Chem., 1998, 37, 1609 CrossRef CAS; (b) H. Sugimoto and Y. Sasaki, Chem. Lett., 1997, 541 CrossRef CAS; (c) H. Sugimoto, M. Kamei, K. Umakoshi, Y. Sasaki and M. Suzuki, Inorg. Chem., 1996, 35, 7082 CrossRef CAS PubMed; (d) N. C. Mösch-Zanetti, S. Köpke, R. Herbst-Irmer and M. Hewitt, Inorg. Chem., 2002, 41, 3513 CrossRef PubMed.
- B. K. Dirghanghi, M. Menon, A. Pramanik and A. Chakravorty, Inorg. Chem., 1997, 36, 1095 CrossRef PubMed.
- I. Chakraborty, S. Bhattacharyya, S. Banerjee, B. K. Dirghangi and A. Chakravorty, J. Chem. Soc., Dalton Trans., 1999, 3747 RSC.
- S. Das, I. Chakraborty and A. Chakravorty, Inorg. Chem., 2003, 42, 6545 CrossRef CAS PubMed.
- P. G. Edwards, J. Jokela, A. Lehtonen and R. Sillanpää, J. Chem. Soc., Dalton Trans., 1998, 3287 RSC.
- (a) C. F. Edwards, W. P. Griffith, A. J. P. White and D. J. Williams, J. Chem. Soc., Dalton Trans., 1992, 957 RSC; (b) J. M. Mayer, Inorg. Chem., 1988, 27, 3899 CrossRef CAS.
- (a) C. J. L. Lock and G. Turner, Can. J. Chem., 1977, 55, 333 CrossRef CAS; (b) L. Hansen, X. Xu, K. T. Tue, Z. Kuklenyik, A. Taylor Jr and L. G. Marzilli, Inorg. Chem., 1996, 35, 1958 CrossRef CAS.
- (a) J. K. Gardner, N. Prariyadath, J. L. Corbin and E. I. Stiefel, Inorg. Chem., 1978, 17, 897 CrossRef CAS; (b) L. A. de-Learie, R. C. Haltiwanger and C. G. Pierpont, Inorg. Chem., 1987, 26, 817 CrossRef CAS.
- A. Mondal, S. Sarkar, D. Chopra, T. N. G. Row and K. K. Rajak, Dalton Trans., 2004, 3, 3244–3250 RSC.
- A. Mondal, S. Sarkar, D. Chopra and K. K. Rajak, Inorg. Chim. Acta, 2006, 359, 2141–2146 CrossRef CAS PubMed.
- S. Basak and K. K. Rajak, Inorg. Chem., 2008, 47, 8813–8822 CrossRef CAS PubMed.
- M. A. L. Marques and E. K. U. Gross, Annu. Rev. Phys. Chem., 2004, 55, 427–455 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, J. Wang, Y. Liu and Y. Yang, J. Am. Chem. Soc., 2009, 131, 8839–8847 CrossRef CAS PubMed.
- X. Gao, Y. Wang, Y. Wang, J. Jia and X. Su, Sci. Sin.: Chim., 2011, 41, 1145–1155 Search PubMed.
- (a) K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251 CrossRef CAS; (b) P. Taehtinen, A. Bagno, K. Klika and K. Pihlaja, J. Am. Chem. Soc., 2003, 125, 4609 CrossRef CAS PubMed; (c) F. Cloran, I. Carmichael and A. S. Serianni, J. Am. Chem. Soc., 2001, 123, 4781 CrossRef CAS PubMed.
- D. E. Grove and G. Wilkinson, J. Chem. Soc., 1966, 1224 RSC.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- (a) M. E. Casida, C. Jamoroski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS PubMed; (b) R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS PubMed; (c) R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS; (b) M. Cossi and V. Barone, J. Chem. Phys., 2001, 115, 4708–4717 CrossRef CAS PubMed; (c) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- (a) T. Liu, H.-X. Zhang and B.-H. Xia, J. Phys. Chem. A, 2007, 111, 8724–8730 CrossRef CAS PubMed; (b) X. Zhou, H.-X. Zhang, Q.-J. Pan, B.-H. Xia and A.-C. Tang, J. Phys. Chem. A, 2005, 109, 8809–8818 CrossRef CAS PubMed; (c) X. Zhou, A.-M. Ren and J.-K. Feng, J. Organomet. Chem., 2005, 690, 338–347 CrossRef CAS PubMed; (d) A. Albertino, C. Garino, S. Ghiani, R. Gobetto, C. Nervi, L. Salassa, E. Rosenverg, A. Sharmin, G. Viscardi, R. Buscaino, G. Cross and M. Milanesio, J. Organomet. Chem., 2007, 692, 1377–1391 CrossRef CAS PubMed.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS PubMed; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS PubMed.
- A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef CAS PubMed.
- (a) K. Wolniski, J. F. Hilton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef; (b) R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS; (c) R. Mcweeny, Phys. Rev., 1962, 126, 1028–1034 CrossRef; (d) F. London, J. Phys. Chem., 1937, 397–409 CAS.
- (a) C. M. Rohlfing, L. C. Allen and R. Ditchfield, Chem. Phys., 1984, 87, 9–15 CrossRef CAS; (b) M. Cossi, V. Barone, B. Mennucci and J. Tomasi, Chem. Phys. Lett., 1998, 286, 253–260 CrossRef CAS; (c) E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef PubMed.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, (Revision A.1), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- SMART; SAINT; SADABS; XPREP; SHELXTL, Bruker AXS Inc., Madison, WI, 1998 Search PubMed.
- G. M. Sheldrick, SHELXTL, v. 6.14, Bruker AXS Inc., Madison, WI, 2003 Search PubMed.
- C. K. Johnson, ORTEP Report ORNL-5138, Oak Ridge National Laboratory, Oak Ridge, TN, 1976 Search PubMed.
- J. Wagler, D. Gerlach and G. Roewer, Inorg. Chim. Acta, 2007, 360, 1935–1942 CrossRef CAS PubMed.
- P. Fita, E. Luzina, T. Dziembowska, D. Kopeć, P. Piątkowski, C. Radzewicz and A. Grabowska, Chem. Phys. Lett., 2005, 416, 305 CrossRef CAS PubMed.
- K. R. Grünwald, G. Saischek, M. Volpe and N. C. Mösch-Zanetti, Inorg. Chem., 2011, 50, 7162–7171 CrossRef PubMed.
- M. M. Abu-Omar and S. I. Khan, Inorg. Chem., 1998, 37, 4979–4985 CrossRef CAS PubMed.
- T. Lis, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 2707–2709 CrossRef.
- B. Machura, R. Kruszynski and M. Jaworska, Polyhedron, 2006, 25, 1111–1124 CrossRef CAS PubMed.
- E. Inego, E. Zangrando, S. Mestroni, G. Fronzoni, M. Stener and E. Alessio, Dalton Trans., 2001, 1338–1346 RSC.
- S. Schmid and J. Strähle, Z. Kristallogr., 1992, 198, 49–59 CrossRef CAS PubMed.
- (a) C. J. L. Lock and G. Turner, Can. J. Chem., 1977, 55, 333 CrossRef CAS; (b) S. Basak and K. K. Rajak, Inorg. Chem., 2008, 47, 8813–8822 CrossRef CAS PubMed; (c) A. Mondal, S. Sarkar, D. Chopra, T. N. Guru Row and K. K. Rajak, Dalton Trans., 2004, 3244–3250 RSC.
- A. Sachse, N. C. Mösch-Zanetti, G. Lyashenko, J. W. Wielandt, K. Most, J. Magull, F. Dall'Antonia, A. Pal and R. Herbst-Irmer, Inorg. Chem., 2007, 46, 7129–7135 CrossRef CAS PubMed.
- A. Schröckeneder, P. Traar, G. Raber, J. Baumgartner, F. Belaj and N. C. Mösch-Zanetti, Inorg. Chem., 2009, 48, 11608–11614 CrossRef PubMed.
- P. Traar, A. Schröckeneder, M. E. Judmaier, F. Belaj, J. Baumgartner, A. Sachse and N. C. Mösch-Zanetti, Eur. J. Inorg. Chem., 2010, 36, 5718–5727 CrossRef.
- W. A. Herrmann, R. W. Fischer and D. W. Marz, Angew. Chem., Int. Ed. Engl., 1991, 30, 1638–1641 CrossRef.
- C. Coperet, H. Adolfsson and B. K. Sharpless, Chem. Commun., 1997, 1565–1566 RSC.
- C. C. Romão, F. E. Kühn and W. A. Herrmann, Chem. Rev., 1997, 97, 3197–3246 CrossRef PubMed.
- F. E. Kühn, M. U. Rauch, G. M. Lobmaier, G. R. J. Artus and W. A. Herrmann, Chem. Ber., 1997, 130, 1427–1431 CrossRef.
- W. A. Herrmann, U. M. Rauch and G. R. J. Artus, Inorg. Chem., 1996, 35, 1988–1991 CrossRef CAS.
- D. W. Pipes and T. J. Meyer, Inorg. Chem., 1986, 25, 3256–3262 CrossRef CAS.
- J. C. Brewer and H. B. Gray, Inorg. Chem., 1989, 28, 3334–3336 CrossRef CAS.
- D. Parker and P. S. Roy, Inorg. Chem., 1988, 27, 4127–4130 CrossRef CAS.
- X. Shan, A. Ellern, I. A. Guzei and J. H. Espenson, Inorg. Chem., 2003, 42, 2362–2367 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic file in CIF format for [ReVO(L1)Cl2], 1; Fig. S1–S7 and Tables S1 and S2. CCDC 1033482. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra15135f |
| This journal is © The Royal Society of Chemistry 2015 |