
Polystyrene resin supported palladium(0) (Pd@PR) nanocomposite mediated regioselective synthesis of 4-aryl-1-alkyl/(2-haloalkyl)-1H-1,2,3-triazoles and their N-vinyl triazole derivatives from terminal alkynes†‡
Arun K. Shilab,
Sandeep Kumarab,
Saurabh Sharmaab,
Abha Chaudharya and
Pralay Das*ab
aNatural Product Chemistry and Process Development, CSIR-Institute of Himalayan Bioresource Technology, Palampur-176061, Himachal Pradesh, India. E-mail: pdas@ihbt.res.in; pdas_nbu@yahoo.com; Fax: +91-1894-230433
bAcademy of Scientific & Innovative Research, New Delhi, India
First published on 8th January 2015
Abstract
An efficient general methodology has been developed for sequential one-pot synthesis of 4-aryl-1-alkyl-1H-1,2,3-triazoles influenced by polystyrene resin supported palladium(0) (Pd@PR) nanocomposite as a heterogeneous catalyst. The present work particularly emphasizes the synthesis of 4-aryl-1-(2-haloalkyl)-1H-1,2,3-triazoles through the selective mono-azidation of 1,2-dihaloethane and subsequent Pd@PR mediated 1,3-dipolar cycloaddition with terminal aryl alkynes. Potassium carbonate promoted dehydrohalogenation of synthesized 4-aryl-1-(2-haloalkyl)-1H-1,2,3-triazoles gave the corresponding N-vinyl derivatives (often used as building blocks for polymers) which are further utilized in the synthesis of 4-aryl-1-(2-arylalkenyl)-1H-1,2,3-triazoles following a Pd@PR catalyzed Heck coupling approach. Furthermore, microwave assisted one pot dehydrochlorination and Heck strategy was adopted to afford 4-phenyl-1-styryl-1H-1,2,3-triazole under Pd@PR catalyzed conditions using iodobenzene as a phenylating agent.
1. Introduction
Substituted 1,2,3-triazoles are ubiquitous structural motifs found in a wide range of potent pharmaceutical compounds and advanced materials (Fig. 1a), and have gained growing interest in diversified research areas.1 In spite of their remarkable metabolic stability, 1,2,3-triazoles have proven amazing medicinal efficacy as non-nucleoside reverse transcriptase inhibitors (Fig. 1b), anti-cancer (Fig. 1c) and anti-HIV (Fig. 1d) agents.2 In recent years, the cutting edge intra as well as trans-disciplinary demands have triggered a major thrust for the development of efficient methodologies for the synthesis of naturally non-occurring 1,2,3-triazole derivatives. The syntheses are mainly based on Huisgen's 1,3-dipolar cycloaddition between organic azides and substituted alkynes.3 But the major challenge relies on the regio-selective control over the thermal [3 + 2] cycloaddition reaction which often gives apparently inseparable mixture of unsymmetrical regioisomers. However, copper4 and ruthenium5 complexes catalyzed azide–alkyne 1,3-dipolar cycloaddition reaction (Cu-AAC and Ru-AAC) were accomplished with improved regioselectivity to procure either 1,4- or 1,5-disubstituted 1,2,3-triazoles exclusively (Scheme 1(A)). Recently, charcoal impregnated Zn was efficiently applied in cycloaddition of arylazides and alkynes to synthesise 1,4-disubstituted 1,2,3-triazoles.6 The transition metal catalyzed regioselective 1,3-dipolar cycloaddition reaction have established the basis of the ‘click chemistry’ and set a wonderful platform for the application in post-synthetic modifications of the 1,2,3-triazole functional scaffolds.1,7 Moreover, inadequate commercial availability of organic azides and the fact that low molecular weight organic azides having low carbon to nitrogen ratio are considered to be potential explosive, have significantly restricted their application in direct azide–alkyne cycloaddition. Ligand aided Pd-catalysts were successfully employed for the synthesis of 1,2,3-triazoles starting from vinyl halides and sodium azide (Scheme 1(A)).8 Nevertheless, the developed homogeneous transition metal catalyzed methodologies bear the inherent drawbacks such as use of costly air sensitive ligands/catalysts, recyclability and metal contamination along with the product. | ||
| Scheme 1 Synthesis of (A) 1,4-disubstituted 1,2,3-triazoles, (B) 1-styryl-1,4-disubstituted 1,2,3-triazoles. | ||
 | ||
| Fig. 1 1,2,3-trizole motif containing value added products. | ||
However, terminal alkynes in presence of heterogeneous palladium are the most unexplored catalytic candidate for azide–alkyne cycloaddition reaction with in situ generated alkyl azides. On the other hand the multistep synthetic approaches have been targeted to derive N-vinyl and N-styryl-1,2,3-triazoles via pyrolytic elimination of polymer supported organo-sulphoxides or selenides.9 Whereas N-vinyl substituted 1,2,3-triazole derivatives are the precursors for industrially important functionalized polymers. The facile polymerization of N-vinyl-1,2,3-triazoles under free radical condition motivated chemists for large scale production of the electron rich polymers.9a,b
As a part of our continuing efforts to broaden the applicability of developed heterogeneous nanocatalysts,10 herein, we describe Pd@PR nanocomposite catalyzed facile sequential one-pot process for the regio-selective synthesis of 4-aryl-1-alkyl-1H-1,2,3-triazoles starting from terminal aryl alkynes, primary alkyl halides and sodium azide. The synthesized 4-aryl-1-(2-haloalkyl)-1H-1,2,3-triazoles were quantitatively converted to the corresponding 4-aryl-1-vinyl-1H-1,2,3-triazoles which further utilized in Heck coupling reaction under the same catalytic condition to afford 4-aryl-1-(2-arylethenyl)-1H-1,2,3-triazoles.
2. Results and discussion
Pd@PR nanocomposite (earlier known as SS-Pd) was previously developed and well characterized in our laboratory.10c–f The straightforward preparation procedure of Pd@PR nanocomposite encompasses in situ reduction of Pd(II) precursor to Pd(0) and their simultaneous deposition over the polystyrene matrix (Experimental section).Initially, the optimization study for azide–alkyne cycloaddition (AAC) reaction among phenyl acetylene, dichloro ethane (DCE) and sodium azide as test substrates was carried out under Pd-catalyzed condition by varying solvents, catalyst loading, temperature and reaction time, and the results were summarized in Table 1. Increasing catalyst loading as well as NaN3 quantity in different solvents led to higher conversion, unlike CH3CN. Among the solvents tested, DMF gave highest yield of the product 1 when employed with Pd@PR (3 mol% Pd) and 3 equiv. of NaN3 with in minimum reaction time 8 h (Table 1, entry 10), and considered as optimized reaction condition. Whereas, Pd(OAc)2 and heterogeneous Pd/C were found to be lesser reactive than Pd@PR. However, catalyst free reaction proceeded to afford traces or lower yield of the product 1 (Table 1, entries 2–4), which further confirmed the role of Pd@PR catalyst for the conversion. The Pd@PR catalyst could be reused up to five times with negligible loss of catalytic activity (first run, 68% yield; fifth run, 58% yield) (ESI†).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Pd-catalyst [mol%] | NaN3 [equiv.] | Solvent | Temp. [°C] | Time [h] | %Yielda |
| a Isolated yields. | ||||||
| 1 | Pd@PR [2] | 2 | 1,4-Dioxane | 90 | 10 | 42 |
| 2 | — | 3 | 1,4-Dioxane | 90 | 12 | Trace |
| 3 | Pd@PR [3] | 3 | (CH2)2Cl2 | 100 | 12 | Trace |
| 4 | — | 3 | DMF | 100 | 8 | 26 |
| 5 | Pd@PR [3] | 3 | 1,4-Dioxane | 100 | 8 | 51 |
| 6 | Pd@PR [2] | 2 | PEG-400 | 100 | 8 | 47 |
| 7 | Pd@PR [3] | 3 | Toluene | 100 | 8 | 53 |
| 8 | Pd@PR [4] | 4 | Toluene | 100 | 8 | 55 |
| 9 | Pd@PR [3] | 2 | CH3CN | 90 | 8 | 38 |
| 10 | Pd@PR [3] | 3 | DMA | 100 | 8 | 58 |
| 11 | Pd@PR [2] | 3 | DMF | 100 | 8 | 62 |
| 12 | Pd@PR [3] | 3 | DMF | 100 | 8 | 68 |
| 13 | Pd@PR [2] | 2 | DMF | 100 | 8 | 52 |
| 14 | Pd@PR [2] | 2 | DMF | 110 | 8 | 60 |
| 15 | Pd(OAc)2 [3] | 3 | DMF | 100 | 10 | 57 |
| 16 | Pd/C [3] | 3 | DMF | 100 | 10 | 53 |

The set reaction condition was further explored for the sequential one-pot [3 + 2] AAC reaction between alkynes and in situ produced alkyl or benzyl azides (Caution! The small molecular weight azides may cause explosion for higher scale reactions) as outlined in Table 2. First, we attempted DCE as alkyl halide source which underwent selective mono-azidation. The aryl alkynes containing both the electron releasing as well as electron withdrawing functional groups gave comparably good yields (61–78%) of the desired 4-aryl-1-(2-chloroethyl)-1H-1,2,3-triazoles 2–8 (Table 2, entries 1–7). The regio-selectivity i.e., 1,4-disubstitution pattern of the product was confirmed by 2D NMR spectral analysis of the product 2 (ESI†). Propargyl benzoate participated smoothly under the standard reaction condition to produce the corresponding 1,2,3-triazole 9 in 73% yield (Table 2, entry 8). Both the heteroaromatic and polyaromatic alkynes were found remarkably reactive for the present PdAAC reaction to afford the desired 4-aryl-1-(2-chloroethyl)-1H-1,2,3-triazoles 10 and 11 in good yields (Table 2, entries 9 and 10). Then we successfully tried 1-bromo-2-chloroethane in place of DCE under the similar reaction condition to obtain 12, 13, and 14 in moderate yields (Table 2, entries 11 to 13) in apparently inseparable mixture of corresponding chloro and bromo derivatives. However formation of higher percentage of chloro-derivatives in all the three cases indicates the expected more prevalent SN2 at C–Br centre. In continuation to our effort to synthesise 4-aryl-1-(2-haloalkyl)-1H-1,2,3-triazoles, we further extended the optimized methodology to achieve 1-(2-hydroxyethyl) and 4-aryl-1-benzyl-1H-1,2,3-triazoles (15–20) in satisfactory yields utilizing 2-bromoethanol and benzyl bromide respectively (Table 2, entries 14 to 19).
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2-X | Products | Time [h] | %Yielda |
| a All are isolated yields; reaction conditions: alkyne (100 mg), dichloroethane (DCE) (8 equiv.), NaN3 (3 equiv.), Pd@PR (3 mol% Pd), DMF, 100 °C.b Reaction conditions: alkyne (100 mg), 1-bromo-2-chloroethane (8 equiv.), NaN3 (3 equiv.), Pd@PR (3 mol% Pd), DMF, 100 °C, the product ratio of chloro and bromo derivatives were calculated on the basis of 1H NMR.c Reaction conditions: alkyne (100 mg), 2-bromoethanol (5 equiv.), NaN3 (3 equiv.), Pd@PR (3 mol% Pd), DMF, 100 °C.d Reaction conditions: alkyne (100 mg), benzylbromide (3 equiv.), NaN3 (3 equiv.), Pd@PR (3 mol% Pd), DMF, 100 °C. | |||||
| 1 | 4-CH3 | (CH2)2Cl2 |  |
8 | 71 |
| 2 | 3-CH3 | (CH2)2Cl2 | 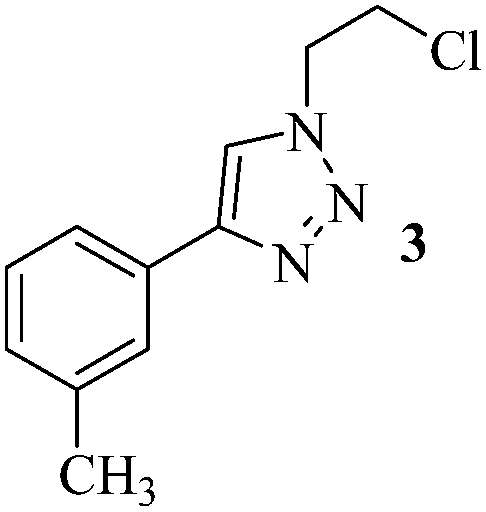 |
9 | 63 |
| 3 | 2-CH3 | (CH2)2Cl2 |  |
9 | 56 |
| 4 | 4-CN | (CH2)2Cl2 |  |
12 | 78 |
| 5 | 4-CF3 | (CH2)2Cl2 |  |
10 | 63 |
| 6 | 4-F | (CH2)2Cl2 |  |
10 | 61 |
| 7 | 2-NH2 | (CH2)2Cl2 |  |
12 | 64 |
| 8 | Benzoyloxy | (CH2)2Cl2 |  |
12 | 73 |
| 9 | 3-thiophene | (CH2)2Cl2 |  |
10 | 59 |
| 10 | 6-methoxy-2-naphthyl | (CH2)2Cl2 | 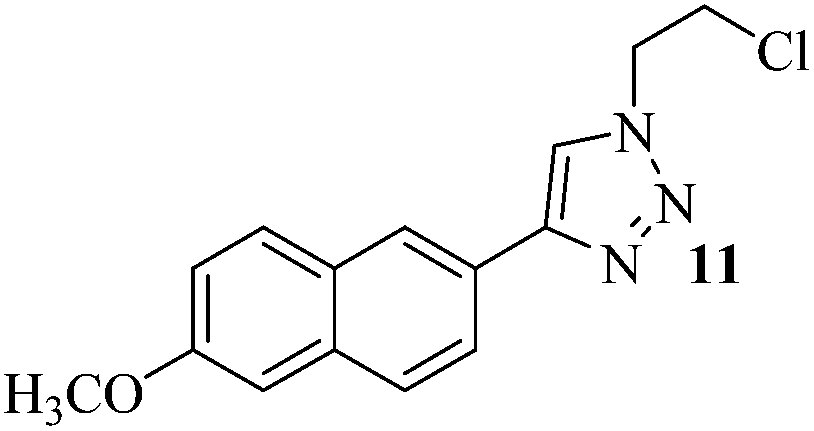 |
12 | 67 |
| 11 | H | (CH2)2ClBr |  |
10 | 63b |
| 12 | 4-CH3 | (CH2)2ClBr |  |
8 | 63b |
| 13 | 6-methoxy-2-naphthyl | (CH2)2ClBr |  |
12 | 66b |
| 14 | H | (CH2)2(OH)Br |  |
10 | 74c |
| 15 | 4-CH3 | (CH2)2(OH)Br |  |
10 | 77c |
| 16 | 4-CN | (CH2)2(OH)Br | 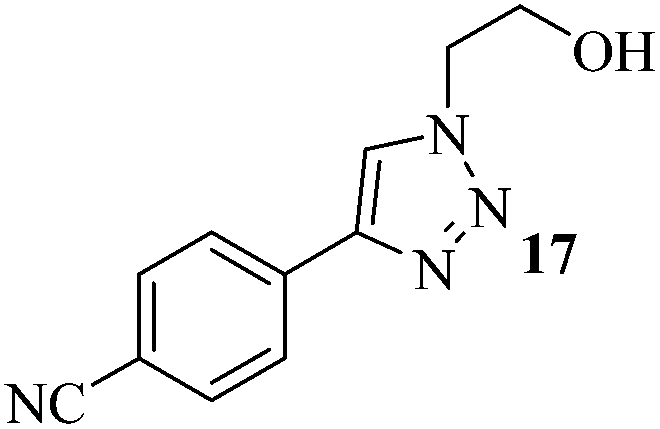 |
10 | 75c |
| 17 | H | PhCH2Br | 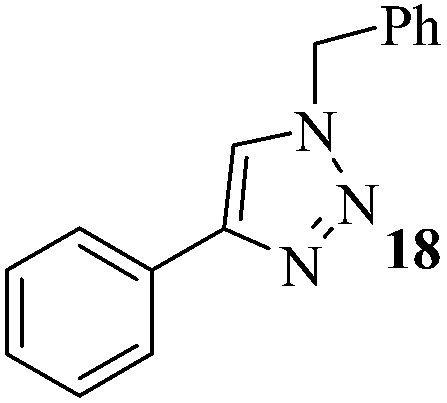 |
10 | 73d |
| 18 | 4-CH3 | PhCH2Br |  |
10 | 74d |
| 19 | 4-CN | PhCH2Br |  |
10 | 70d |

Mechanistically it is presumed that both the pathways A and B (Scheme 2) might follow to give the desired product VI. Surface bound Pd–alkyne π-complex II collapses to form σ-complex III which could lead through either pathway A or pathway B analogous to the reported similar catalytic cycles.4b These pathways are distinguished by their earlier (pathway A) and later (pathway B) substitution reaction (SN2) step involving organo halide. However, treatment of commercially available 4-phenyl-1H-1,2,3-triazole with 1,2-dichloroethane under the similar reaction condition did not produce 1-(2-chloroethyl)-4-phenyl-1H-1,2,3-triazole 1, which ruled out pathway A.
 | ||
| Scheme 2 Plausible mechanistic pathways for Pd-catalyzed azide–alkyne cycloaddition reaction. | ||
The reported tedious synthetic procedures11 for N-vinyl-1H-1,2,3-triazoles have urged us to develop an alternative route (Table 3). Here we utilized the synthesized 4-aryl-1-(2-haloethyl)-1H-1,2,3-triazoles (Table 2) in milder base K2CO3 promoted facile dehydrochlorination to obtain corresponding 4-aryl-1-ethenyl-1H-1,2,3-triazoles (21–29) in good to excellent yields (Table 3).
|
|---|
| a All are isolated yields; reaction conditions: 4-aryl-1-(2-chloroethyl)- 1H-1,2,3-triazole (100 mg), K2CO3 (2 equiv.), DMF (2 mL), 110 °C, 3 h. |
 |

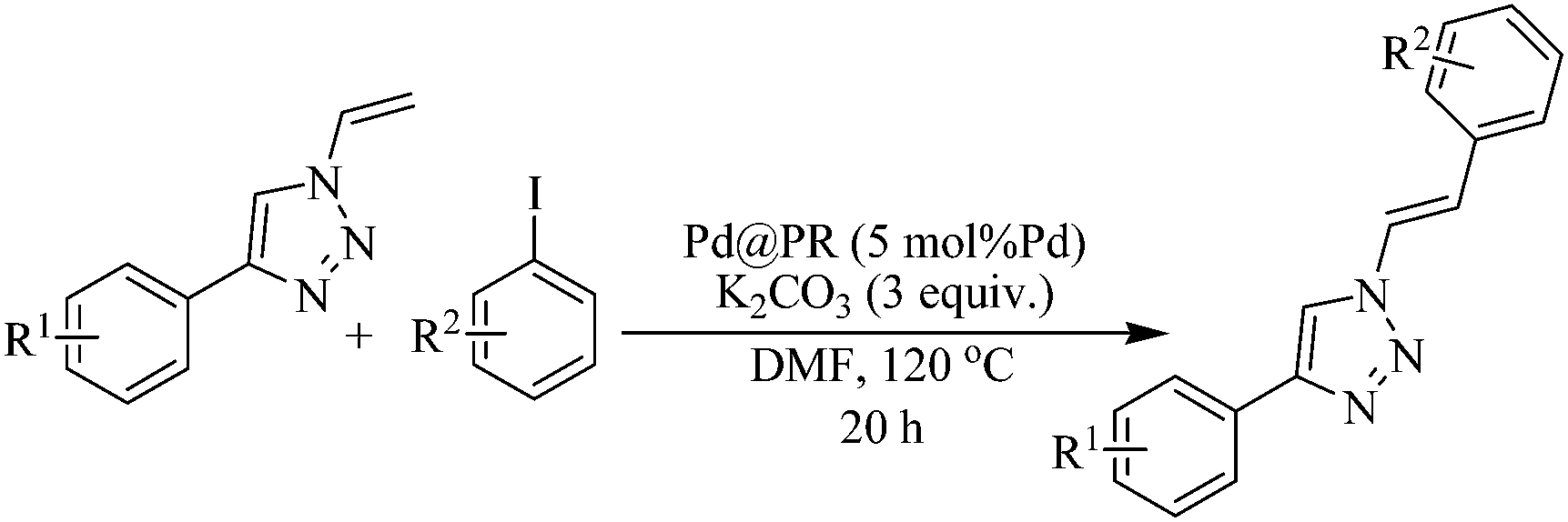
Thus the synthesized N-vinyl-1H-1,2,3-triazole derivatives further open up a spurring opportunity for us to develop an alternative method of Heck coupling strategy to synthesize 4-aryl-1-(2-arylethenyl)-1H-1,2,3-triazoles employing Pd@PR catalyst (Table 3). Whereas, in previous reports costly organo S or Se precursors and tiresome procedures greatly obstructed their synthesis (Scheme 1(B)).12 The differently substituted electron rich aryl iodides easily participated in the set protocol of Heck coupling reaction with 4-phenyl-1-vinyl-1H-1,2,3-triazole following regio-selective terminal arylation to furnish moderate to good yields of the products 30–37 (Table 4) with high E selectivity (confirmed by 1H NMR).
|
|---|
| a All are isolated yields; reaction conditions: 4-aryl-1-vinyl-1,2,3-triazole (100 mg), aryl iodide (1.5 equiv.), Pd@PR (5 mol% Pd), K2CO3 (3 equiv.), DMF (2 mL), 120 °C, 20 h; E/Z ratio was calculated on the basis of 1H NMR. |
 |
Further, we also targeted a sequential one-pot approach for dehydro-halogenation and Heck coupling on 1-(2-chloroethyl)-4-phenyl-1H-1,2,3-triazole (1) with iodobenzene under microwave irradiation to procure 4-phenyl-1-styryl-1H-1,2,3-triazole 30 in 51% yield (Scheme 3).
 | ||
| Scheme 3 Microwave assisted one-pot synthesis of 4-phenyl-1-vinyl-1H-1,2,3-triazole. | ||
3. Conclusion
In summary, we have developed Pd@PR nanocomposite catalyzed general single pot strategy for azide–alkyne cyloaddition (AAC) reaction to synthesise 1,4-disubstituted-1,2,3-triazoles in a regio-selective manner. The developed unprecedented PdAAC approach was specially moved towards 4-aryl-1-(2-haloethyl)-1H-1,2,3-triazole derivatives synthesis. However, other N-alkyl, benzyl substituted surrogates were also procured either by extension or slight variation of the standard protocol. The prepared 4-aryl-1-(2-chloroethyl)-1H-1,2,3-triazoles were utilized in a facile K2CO3 promoted dehydrochlorination to obtain the corresponding N-vinyl derivatives as industrially consumable polymer precursor. The terminally vacant vinyl functionality were successfully utilized for Pd@PR catalysed Heck coupling with aryl iodides to give a series of 4-aryl-1-(2-arylethenyl)-1H-1,2,3-triazole derivatives. In continuation Pd@PR catalysed microwave assisted one pot dehydrohaloginative Heck coupling also opened a new scope to access N-vinyl-1H-1,2,3-triazole analogues. The combined results provide an outstanding example of catalytic efficiency and selectivity of Pd@PR nanocomposite, and could find academic as well as industrial interest.4. Experimental section
General methods
Reagents of high quality were purchased from Sigma Aldrich and Loba Cheime. Amberlite® IRA 900 Cl− resin used as solid support was purchased from Acros Organics. Silica gel (60–120 mesh size) for column chromatography was procured from Sd Fine-chem Ltd. Commercial reagents and solvents were of analytical grade and were purified by standard procedures prior to use. Thin layer chromatography was performed using pre coated silica gel plates 60 F254 (Merck) in UV light detector. Some experiments were performed on CEM Discover focused microwave (2450 MHz, 300 W). The temperature of reactions in monomode microwave experiments was measured by an inbuilt infrared temperature probe that determined the temperature on the surface of reaction flask. The sensor is attached in a feedback loop with an on-board microprocessor to control the rate of temperature rise. All the melting points are uncorrected and were determined on a Barnstead Electrothermal 9100 capillary melting point apparatus. ESIMS spectra were determined using Waters Micromass Q-TOF Ultima Spectrometer. 1H and 13C NMR spectra were recorded using a Bruker Avance 600 spectrometer operating at 600 MHz (1H) and 150 MHz (13C). Spectra were recorded at 25 °C in CDCl3 [residual CHCl3 (δH 7.26 ppm) or CDCl3 (δC 77.00 ppm) and MeOD (δH 3.30, 4.78 ppm) or (δC 49.0 ppm) [residual (as international standard)] with TMS as internal standard. Chemical shifts were calculated according to the residual solvents' standard peaks. Chemical shifts were recorded in δ (ppm) relative to the TMS and NMR solvent signal, coupling constants (J) are given in Hz and multiplicities of signals are reported as follows: s, singlet; d, doublet; dd, double doublet; t, triplet; q, quartet; m, multiplet.Preparation of Pd@PR nanocomposite
The solution of 25 mg of NaBH4 in 10 mL of water (0.065 (M) solution) was added to 1 g of polystyrene resin (Amberlite IRA 900 Cl− form) in a 25 mL round bottom flask. The mixture was stirred for 4 h at room temperature. Then the resin was washed with water till the pH became neutral and then with acetone to remove water from the solid surface. The partially borohydride exchanged resin beads were dried under reduced pressure. The dried borohydride exchanged resin beads were added into the warm (100 °C) solution of palladium acetate (10 mg) in dry DMF (3 mL) and the mixture was stirred for 1 h or till the brown colored solution changed to colorless and simultaneously white solid beads turned black. After cooling, the beads were filtered through a cotton bed, washed with water and acetone, and dried under reduced pressure.General experimental procedure for 4-aryl-1-(2-haloethyl)-1H-1,2,3-triazole (1–20) synthesis
A mixture of aryl alkyne (100 mg), sodium azide (3 equiv.), 1,2-dihaloethane (8 equiv.) and Pd@PR (3 mol% Pd) were taken in an oven dried 40 mL reaction vial with screw cap. Equal volume (as compared to 1,2-dihaloethane) of dry DMF was added into it. The reaction mixture was then stirred under nitrogen in a pre heated oil bath of temperature 100 °C. Progress of the reaction was monitored by TLC. On completion, the cooled reaction mixture was extracted with ethyl acetate (3 × 5 mL) by addition of 2 mL of water and dried over anhydrous Na2SO4. Evaporation of the combined organic layer followed by column chromatography (hexane–ethyl acetate gradient) over silica gel (mesh 60–200) afforded 4-aryl-1-(2-haloethyl)-1H-1,2,3-triazoles (1–14). Products 15–20 were also synthesised applying similar procedure.General experimental procedure for 4-aryl-1-vinyl-1H-1,2,3-triazole (21–29) synthesis
To a mixture of 4-aryl-1-(2-haloethyl)-1H-1,2,3-triazole (100 mg) and K2CO3 (2 equiv.) in an oven dried 40 mL reaction vial was added 2 mL of dry DMF. The reaction mixture was then stirred under nitrogen in a pre heated oil bath of temperature 110 °C for 3 hours. Progress of the reaction was monitored by TLC. On completion, the cooled reaction mixture was extracted with ethyl acetate (3 × 3 mL) by addition of 2 mL of water and dried over anhydrous Na2SO4. Evaporation of the combined organic layer followed by column chromatography (hexane–ethyl acetate gradient) over silica gel (mesh 60–200) afforded 4-aryl-1-vinyl-1H-1,2,3-triazoles (21–29).General experimental procedure for 4-aryl-1-(2-arylvinyl)-1H-1,2,3-triazoles (30–37) synthesis
To a mixture of 4-aryl-1-vinyl-1H-1,2,3-triazole (100 mg), aryl iodide (1.5 equiv.), K2CO3 (2 equiv.) and Pd@PR (5 mol% Pd) in an oven dried 40 mL reaction vial was added 2 mL of dry DMF. The reaction mixture was then stirred under nitrogen in a pre heated oil bath of temperature 120 °C for 20 hours. Progress of the reaction was monitored by TLC. On completion, the cooled reaction mixture was extracted with ethyl acetate (3 × 5 mL) by addition of 2 mL of water and dried over anhydrous Na2SO4. Evaporation of the combined organic layer followed by column chromatography (hexane–ethyl acetate gradient) over silica gel (mesh 60–200) afforded 4-aryl-1-(2-arylvinyl)-1H-1,2,3-triazoles (30–37).Typical experimental procedure for the one pot synthesis of 4-phenyl-1-styryl-1H-1,2,3-triazole (30) synthesis
A mixture of 1-(2-chloroethyl)-4-phenyl-1H-1,2,3-triazole (100 mg, 0.483 mmol), iodobenzene (147.69 mg, 0.724 mmol), K2CO3 (200 mg, 1.449 mmol) and Pd@PR (324 mg, 5 mol% Pd) in 2 mL of dry DMF was irradiated with focused MW at 120 °C (100 W, 100 psi) for 45 min in a closed vessel equipped with automated pressure device. On completion, the cooled reaction mixture was extracted with ethyl acetate (3 × 5 mL) by addition of 2 mL of water and dried over anhydrous Na2SO4. Evaporation of the combined organic layer followed by column chromatography (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethylacetate = 90:10) over silica gel (mesh 60–200) afforded 30 as white solid (73.67 mg, 51%). 1H NMR (600 MHz, CDCl3) δ 7.18–7.20 (d, J = 14.4 Hz, 1H), 7.35–7.50 (m, 9H), 7.80–7.83 (d, J = 15 Hz, 1H), 7.88–7.90 (d, J = 8.4 Hz, 2H), 8.09 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 116.48, 121.57, 123.07, 125.88 (2C), 126.72 (2C), 128.51, 128.80, 128.92, 129.04 (2C), 130.04, 133.58, 148.04. ESIMS data: m/z calcd for [M + H]+ C16H14N3 248.1187, obsd. 248.1178.
:ethylacetate = 90:10) over silica gel (mesh 60–200) afforded 30 as white solid (73.67 mg, 51%). 1H NMR (600 MHz, CDCl3) δ 7.18–7.20 (d, J = 14.4 Hz, 1H), 7.35–7.50 (m, 9H), 7.80–7.83 (d, J = 15 Hz, 1H), 7.88–7.90 (d, J = 8.4 Hz, 2H), 8.09 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 116.48, 121.57, 123.07, 125.88 (2C), 126.72 (2C), 128.51, 128.80, 128.92, 129.04 (2C), 130.04, 133.58, 148.04. ESIMS data: m/z calcd for [M + H]+ C16H14N3 248.1187, obsd. 248.1178.
Acknowledgements
Authors are grateful to Director CSIR-IHBT for providing necessary facilities during the course of work. The authors thank CSIR, New Delhi for financial support as part of XII Five Year Plan programme under title ORIGIN (CSC-0108). AKS, SK, SS and AC thank CSIR and UGC, New Delhi for awarding fellowships.Notes and references
- (a) A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388–1405 RSC; (b) C. O. Kappe and E. Van der Eycken, Chem. Soc. Rev., 2010, 39, 1280–1290 RSC; (c) M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed; (d) H. Nandivada, X. Jiang and J. Lahann, Adv. Mater., 2007, 19, 2197–2208 CrossRef CAS; (e) J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed; (f) K. D. Hanni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240–1251 RSC.
- (a) A. Brik, J. Muldoon, Y.-C. Lin, J. H. Elder, D. S. Goodsell, A. J. Olson, V. V. Fokin, K. B. Sharpless and C.-H. Wong, ChemBioChem, 2003, 4, 1246–1248 CrossRef CAS PubMed; (b) M. J. Soltis, H. J. Yeh, K. A. Cole, N. Whittaker, R. P. Wersto and E. C. Kohn, Drug Metab. Dispos., 1996, 24, 799–806 CAS; (c) R. Alvarez, S. Velazquez, A. San-Felix, S. Aquaro, E. De Clercq, C.-F. N. Perno, A. Karlsson, J. Alzarini and M. J. Camarasa, J. Med. Chem., 1994, 37, 4185–4194 CrossRef CAS.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 (Angew. Chem., 1963, 75, 604–637) CrossRef.
- (a) B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed; (b) L. Ackermann and H. K. Potukuchi, Org. Biomol. Chem., 2010, 8, 4503–4513 RSC; (c) S. D. Gonzlez, A. Correa, L. Cavallo and S. P. Nolan, Chem.–Eur. J., 2006, 12, 7558–7564 CrossRef PubMed.
- (a) J. R. Johansson, P. Lincoln, B. Norden and N. Kann, J. Org. Chem., 2011, 76, 2355–2359 CrossRef CAS PubMed; (b) P. N. Liu, H. X. Siyang, L. Zhang, S. K. S. Tse and G. Jia, J. Org. Chem., 2012, 77, 5844–5849 CrossRef CAS PubMed.
- X. Meng, X. Xu, T. Gao and B. Chen, Eur. J. Org. Chem., 2010, 75, 5409–5414 CrossRef.
- J. M. Holub and K. Kirshenbaum, Chem. Soc. Rev., 2010, 39, 1325–1337 RSC.
- (a) J. Barluenga, C. Valdés, G. Beltrán, M. Escribano and F. Aznar, Angew. Chem., Int. Ed., 2006, 45, 6893–6896 CrossRef CAS PubMed; (b) W. Zhang, C. Kuang and Q. Yang, Synthesis, 2010, 283–287 Search PubMed; (c) S. Kamijo, T. Jin, Z. Huo and Y. Yamamoto, Tetrahedron Lett., 2002, 43, 9707–9710 CrossRef CAS.
- (a) S. Beghdadi, I. A. Miladi, D. Addis, H. B. Romdhane, J. Bernarda and E. Drockenmuller, Polym. Chem., 2012, 3, 1680–1692 RSC; (b) V. N. Kizhnyaev, F. A. Pokatilov, N. A. Tsypina, G. V. Ratovskii, L. I. Vereshchagin and A. I. Smirnov, Russ. J. Org. Chem., 2002, 38, 1056–1057 CrossRef CAS; (c) Q.-Y. Wang, W.-S. Sheng, S.-R. Sheng, Y. Li and M.-Z. Cai, Synth. Commun., 2014, 44, 59–67 CAS.
- (a) N. R. Guha, C. B. Reddy, N. Aggarwal, D. Sharma, A. K. Shil, Bandna and P. Das, Adv. Synth. Catal., 2012, 354, 2911–2915 CrossRef CAS; (b) A. K. Shil and P. Das, Green Chem., 2013, 15, 3421–3428 RSC; (c) P. Das, D. Sharma, A. K. Shil and A. Kumari, Tetrahedron Lett., 2011, 52, 1176–1178 CrossRef CAS PubMed; (d) A. K. Shil, D. Sharma, N. R. Guha and P. Das, Tetrahedron Lett., 2012, 53, 4858–4861 CrossRef CAS PubMed; (e) A. K. Shil, N. R. Guha, D. Sharma and P. Das, RSC Adv., 2013, 3, 13671–13676 RSC; (f) C. B. Reddy, A. K. Shil, N. R. Guha, D. Sharma and P. Das, Catal. Lett., 2014, 144, 1530–1536 CrossRef PubMed; (g) Bandna, N. Aggarwal and P. Das, Tetrahedron Lett., 2011, 52, 4954–4956 CrossRef CAS PubMed; (h) Bandna, N. R. Guha, A. K. Shil, D. Sharma and P. Das, Tetrahedron Lett., 2012, 53, 5318–5322 CrossRef CAS PubMed; (i) D. Sharma, S. Kumar, A. K. Shil, N. R. Guha, Bandna and P. Das, Tetrahedron Lett., 2012, 53, 7044–7051 CrossRef CAS PubMed.
- (a) K. Dabak and A. Akar, Heterocycl. Commun., 2011, 8, 61–64 Search PubMed; (b) L. I. Vereshchagin, L. G. Tikhonova, A. V. Maksikova, L. D. Gavrilov and G. A. Gareev, Zh. Org. Khim., 1979, 15, 612–618 CAS; (c) V. N. Kizhnyaev, F. A. Pokalitov, N. A. Tsypina, G. V. Ratovski, L. I. Vereshchagin and A. I. Smirnov, Russ. J. Org. Chem., 2002, 38, 1056–1059 CrossRef CAS; (d) D. Sikora and T. Gajda, Tetrahedron, 1998, 54, 2243–2250 CrossRef CAS; (e) H. Nulwala, D. J. Burke, A. Khan, A. Serrano and C. J. Hawker, Macromolecules, 2010, 43, 5474–5477 CrossRef CAS.
- (a) J. H. Boyer, W. E. Krueger and R. Modler, J. Org. Chem., 1969, 34, 1987–1989 CrossRef CAS; (b) H.-Y. Su, S.-R. Sheng, X.-L. Zhang, X.-Y. Ding and M.-Z. Cai, Phosphorus, Sulfur Silicon Relat. Elem., 2013, 188, 1591–1598 CrossRef CAS; (c) J.-W. Jiang, S.-R. Sheng, X.-L. Zhang, Z. Fang and M.-Z. Cai, Synth. Commun., 2013, 43, 2784–2792 CrossRef CAS; (d) M. Kavitha, B. Mahipal, P. S. Mainkar and S. Chandrasekhar, Tetrahedron Lett., 2011, 52, 1658–1662 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Graphical representation of Pd@PR catalyst preparation, recyclability experiment, typical experimental procedures, 2D NMR spectral analysis of product 2 and copies of 1H NMR, 13C NMR, and ESIMS spectra. See DOI: 10.1039/c4ra15133j |
| ‡ IHBT Communication no. 3757. |
| This journal is © The Royal Society of Chemistry 2015 |