
Investigations of clustering of ions and diffusivity in concentrated aqueous solutions of lithium chloride by molecular dynamic simulations†
Abstract
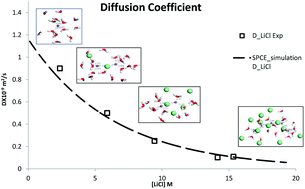
The interactions between lithium (Li+) ions, chloride (Cl−) ions and water molecules in aqueous LiCl solutions and their effect on the dynamic and equilibrium properties of the salt solutions have been investigated by molecular dynamics (MD) simulations. The optimized potentials for liquid simulations for all atoms (OPLS-AA) force field have been used to study various properties of lithium chloride solutions for the concentrations, in the range of 0.1 M to 19.28 M. The MD simulation with the OPLS-AA force field gives a fair explanation of many important properties of alkali salt solutions which are in agreement with the experimental results. A microscopic description of LiCl solutions and diffusivity of LiCl obtained by simulation are in good agreement with the experimental data. The MD simulation indicated a strong solvation of monovalent ions in water and cluster formation of the cations at higher salt concentrations. The diffusion coefficient of LiCl decreases depending on the coordination structure of ions that changes with the salt concentration.
 Please wait while we load your content...
Please wait while we load your content...

