DOI:
10.1039/C4RA15070H
(Paper)
RSC Adv., 2015,
5, 16319-16327
Electrochemical performance of an asymmetric supercapacitor based on graphene and cobalt molybdate electrodes
Received
23rd November 2014
, Accepted 29th January 2015
First published on 29th January 2015
Abstract
In this article, we report the fabrication and electrochemical performance of asymmetric supercapacitors (ASCs) based on a reduced graphene oxide (rGO) negative electrode and a cobalt molybdate (CoMoO4) positive electrode. The rGO and CoMoO4 electrode materials were synthesized by hydrothermal and sonochemical methods, respectively. Physico-chemical characterization techniques such as X-ray diffraction, field-emission scanning electron microscopy, Fourier-transform infrared spectroscopy, Raman spectroscopy, and nitrogen adsorption–desorption isotherm analysis were used to characterize the electrode materials. The rGO nanosheets and CoMoO4 nanostructures delivered a specific capacitance of about 168.8 and 98.34 F g−1, respectively measured in a three electrode system. The rGO‖CoMoO4 ASC device demonstrated a maximum specific capacitance of 26.16 F g−1 (at a current density of 0.5 mA cm−2), an energy density of 8.17 W h kg−1, and a maximum working voltage of 1.5 V. The fabricated device possessed excellent capacitance retention of about 85% after 4000 cycles (at a current density of 1.0 mA cm−2) suggesting the superior cyclic stability of the fabricated ASC device.
1. Introduction
In recent years, there is an increasing demand for environmental friendly, small, lightweight, flexible energy storage and conversion systems (e.g., batteries, fuel cells, and capacitors) have attracted a great attention for portable electronics (e.g., computers and mobile phones) in the fast-growing market.1–3 Supercapacitors (SCs), also known as electrochemical capacitors or ultracapacitors, are directed towards achieving high specific energy, high specific power, long cycle life, etc., at relatively low cost, and they store energy via (i) electrical double layer capacitance or (ii) pseudocapacitance. The electrical double layer capacitance arises due to the reversible electrolyte ions adsorbed at the electrode/electrolyte interface whereas the pseudocapacitance originates due to the occurrence of redox reactions at the electrode surface.4–6 However, in comparison with rechargeable batteries, SCs are limited by a low energy density. Thus, an essential target for SCs is to improve the energy density of the SC, while retaining a high power density and long cycle life. According to the equation E = 0.5CV2, where E is the energy density, C is the capacitance, and V is the cell voltage, the energy density of SCs can be enhanced by increasing the capacitance and cell voltage.7,8 Two strategies have been investigated to increase the potential window, (1) the use of organic electrolytes instead of aqueous electrolytes and (2) the use of asymmetric SCs (ASCs). The system using organic electrolytes shows low capacitive performance and low power capability due to limited ion concentration and ionic conductivity.9 In contrast, the enhancement in the potential window displayed by ASCs has been impressive, resulting in higher energy density in aqueous electrolytes. ASCs commonly have an electrochemical double-layer (capacitor-type) electrode created from carbon materials and a second electrode constructed from a pseudocapacitive (battery-type) material; together, the two components allow the ASC to exhibit two different potential windows in the same electrolyte.10–12 Thus, ASCs are configured to fully utilize the different potential windows of the two electrodes to maximize the operation voltage of the cell system.13
Recently, considerable research effort has been devoted to increasing the working voltage levels of various ASCs using different materials for the negative and positive electrodes.13–15 In these studies, the negative electrode is mainly composed of carbonaceous materials, such as activated carbon, carbon nanotubes (CNTs), and graphene.16–18 Among these, graphene is an ultrathin two-dimensional hexagonal lattice of sp2 carbon atoms, covalently bonded along two plane directions.19 Graphene has been investigated extensively for its electrical, electrochemical, and electronic applications due to its high strength, as well as its excellent thermal and electrical conductivity.20,21 The exceptional electrochemical behavior of graphene can be explained by its high specific surface area, providing the electrolyte ions easy access to the electrode.22 In addition, graphene-based materials can be easily fabricated by simple chemical synthesis of graphite oxide. Besides, graphene has been extensively studied as a negative electrode material for ASCs.18,21,23,24 Hitherto, metal oxides/hydroxides, binary metal oxides, and conducting polymers, among other materials, have been investigated for use as a positive electrode material for ASCs.23–27 Among them, binary metal oxides/hydroxides are considered to be especially effective due to their multiple oxidation states and high electrical conductivities.28,29 Recently, metal molybdates, such as CoMoO4, NiMoO4, and MnMoO4, have demonstrated enhanced electrochemical performance over the single-component oxides.29–31 In particular, cobalt molybdate (CoMoO4) SCs exhibit a high electrochemical performance, high rate capability, and high durability,32 in addition to being nontoxic, environmentally safe, and inexpensive.
Several studies have investigated on CoMoO4-based ASCs. Liu et al. fabricated an ASC based on CoMoO4–NiMoO4·xH2O as the positive electrode and activated carbon as the negative electrode,33 while Senthilkumar et al. synthesized an ASC using β-NiMoO4–CoMoO4·xH2O as the positive electrode and activated carbon as the negative electrode.34 In this article, we report an ASC fabricated using CoMoO4 nanostructures as the positive electrode and reduced graphene oxide nanosheets as the negative electrode material, with 1.0 M sodium hydroxide (NaOH) used as the electrolyte. Commercial filter paper was used as a separator. This ASC device achieved a specific capacitance of 26.16 F g−1 and an energy density of 8.17 W h kg−1 at a constant discharge current density of 0.5 mA cm−2 for an operational voltage of 1.5 V.
2. Experimental section
2.1. Materials and methods
Sodium molybdate (Na2MoO4) and graphite powder were purchased from Sigma-Aldrich Ltd, South Korea. Potassium permanganate (KMnO4), sulfuric acid (H2SO4), hydrogen peroxide (H2O2), NaOH, hydrochloric acid (HCl), cobalt chloride hexahydrate (CoCl2·6H2O), methanol (CH3OH), and ethanol (C2H5OH) were purchased from Daejung Chemicals Ltd, South Korea. All chemicals used in this research were of research grade; double-distilled water was used throughout the experiment. Ultrasound irradiation was carried out with a SONIX VCX 750 system (20 kHz; 750 W) using a direct immersion titanium horn.
2.2. Preparation of rGO nanosheets
The rGO nanosheets were prepared using graphene oxide (GO) (synthesized via the modified Hummers method35) as the starting material. A hydrothermal method was used to reduce GO into rGO nanosheets, with water as the solvent. Briefly, 80 mg of GO was dispersed in water and sonicated in a bath-type sonicator for 1 h to obtain a uniform brown-colored GO dispersion. The solution was then transferred to a 100 mL Teflon autoclave, covered by a stainless steel reactor, and kept at a constant temperature of 150 °C for 10 h. After the reaction, the Teflon was cooled to room temperature naturally; the color of the suspension turned black, indicating the reduction of GO into rGO. The rGO suspension was washed thoroughly and centrifuged several times with distilled water and ethanol to remove the residual ions. Finally, the sample was dried at 60 °C for 5 h in a hot-air oven.
2.3. Preparation of CoMoO4 nanostructures
The CoMoO4 nanostructures were synthesized via a sonochemical approach using Na2MoO4 and CoCl2 as the starting precursors, as reported in our previous study.36 Briefly, 1 M of CoCl2·6H2O in CH3OH and 1 M of Na2MoO4 in water were prepared in two separate beakers. The CoCl2 solution underwent ultrasound irradiation, followed by dropwise addition of the Na2MoO4 solution. Note that during this process, the entire solution was stirred gently with a Teflon-coated magnetic stir bar for 1.5 h to obtain violet-colored precipitates of CoMoO4·xH2O. The precipitate was thoroughly washed with distilled water and ethanol until the impurities and residual ions were eliminated from the final product. Lastly, the CoMoO4·xH2O precipitate was allowed to dry at 100 °C for 3 h, followed by calcination at 500 °C for 3 h, resulting in the formation of CoMoO4 nanostructures.
2.4. Materials characterization
X-ray diffraction (XRD) analysis was performed using an X-ray diffractometer system (D/MAX 2200H, Bede 200, Rigaku Instruments C), operating at 40 kV and 40 mA with Cu-Kα radiation. The surface morphology of the prepared materials was evaluated by field emission-scanning electron microscopy (FE-SEM, JSM-6700F, JEOL). Fourier-transform infrared (FTIR) spectra were obtained with a Thermo Scientific Nicolet-6700 FT-IR spectrometer using pure KBr as the background. The sample was mixed with KBr, and the mixture was then dried and pressed into transparent pellets for measurement. Raman spectroscopy was performed on the samples using a LabRam HR Evolution Raman spectrometer (Horiba Jobin-Yvon, France). The Raman system used an Ar+ ion laser operating at a laser power of 10 mW and an excitation wavelength of 514 nm; a 10 s data-point acquisition time was used. The N2 adsorption–desorption isotherms of the samples were measured at 77 K using a NOVA 2000 system (Quantachrome, USA).
2.5. Preparation of the working electrodes and electrochemical studies
The working electrodes were prepared using the following procedure. The active material (CoMoO4 or rGO) was ground with carbon black and polyvinylidene difluoride (PVDF; mixture ratio: 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:5) using N-methyl pyrrolidone (NMP). A stainless steel substrate (1 × 1 cm2) was then coated with the mixture, and the coated substrate was allowed to dry at 80 °C overnight. The mass loading of the rGO nanosheets and CoMoO4 nanostructures on the electrode was 1.8 and 1.1 mg respectively, and a solution containing 1 M NaOH was used as the electrolyte. The electrochemical behavior of the positive and negative electrodes was examined at room temperature using a three-electrode system: CoMoO4 or rGO as the working electrode, silver/silver chloride (Ag/AgCl) as the reference electrode, and platinum as the counter electrode. Finally, ASCs were fabricated in the form of a sandwich-type electrode, with an electrolyte-immersed filter paper used as the separator. All of the electrochemical properties of the ASCs were examined via cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) measurements using an Autolab PGSTAT302N electrochemical workstation.
:20:5) using N-methyl pyrrolidone (NMP). A stainless steel substrate (1 × 1 cm2) was then coated with the mixture, and the coated substrate was allowed to dry at 80 °C overnight. The mass loading of the rGO nanosheets and CoMoO4 nanostructures on the electrode was 1.8 and 1.1 mg respectively, and a solution containing 1 M NaOH was used as the electrolyte. The electrochemical behavior of the positive and negative electrodes was examined at room temperature using a three-electrode system: CoMoO4 or rGO as the working electrode, silver/silver chloride (Ag/AgCl) as the reference electrode, and platinum as the counter electrode. Finally, ASCs were fabricated in the form of a sandwich-type electrode, with an electrolyte-immersed filter paper used as the separator. All of the electrochemical properties of the ASCs were examined via cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) measurements using an Autolab PGSTAT302N electrochemical workstation.
3. Results and discussion
The crystalline nature of the synthesized materials was investigated by XRD analysis (Fig. 1). According to XRD pattern of Fig. 1A, the prepared GO exhibited a sharp dominant peak at 10.43° (with an interlayer spacing of 0.847 nm), which was assigned to (001) reflection. After hydrothermal reduction of GO, these peaks disappeared completely; a strong, broad peak appeared at a 2θ value of 24.29° (with an interlayer spacing of 0.365 nm), which was assigned to the (002) plane of the graphitic domains due to the re-graphitization process.37,38 Fig. 1B shows the diffraction pattern for monoclinic CoMoO4. The maximum intensity was observed at 2θ = 26.43°, corresponding to the (002) plane. All of the diffraction peaks matched exactly the standard patterns for monoclinic CoMoO4, as determined by the Joint Committee on Powder Diffraction Standards (card no. 21-0868). The calculated interlayer spacing of CoMoO4 (002) was ∼0.336 nm, in good agreement with the value reported by Mai et al.30 FE-SEM was used to examine the morphology of the prepared materials (Fig. 2). The low and high magnification images of hydrothermally synthesized rGO is shown in Fig. 2A and B, respectively, exhibited a sheet-like structure of graphene. The inset of Fig. 2A displaying the image of GO, it depicts that wrinkled and folded sheet like form. Fig. 2C and D display low and high magnification images of sonochemically synthesized CoMoO4, respectively; a plate-like structure was revealed, approximately several hundreds of nanometers in size.
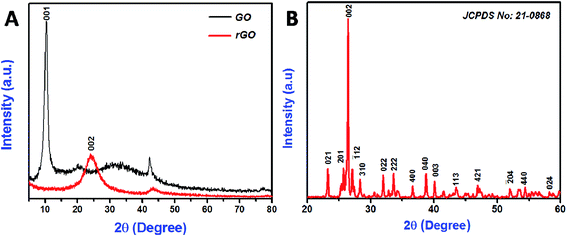 |
| | Fig. 1 XRD pattern of the synthesized GO and rGO nanosheets (A), CoMoO4 nanostructures (B). | |
 |
| | Fig. 2 FE-SEM image of rGO nanosheets at low (A) and high magnifications (B), GO (inset of (A)), CoMoO4 nanostructures at low (C) and high magnifications (D). | |
The bonding nature of the synthesized CoMoO4 nanostructures and rGO nanosheets are examined by FTIR analysis (Fig. 3A and B). Three major bands were observed in the CoMoO4 spectra (Fig. 3A) at 948, 641, and 441 cm−1, corresponding to the vibrational mode of distorted MoO4, vibrational mode of Mo–O, and vibrations due to the Co and Mo building blocks of CoMoO4, respectively.36 The peak at 1638 cm−1 indicates that the presence of physically adsorbed water molecule on the surface of the CoMoO4.39 The removal of oxygen-containing functional groups of GO after hydrothermal reduction was well examined in Fig. 2B. In GO, the oxygenated functional groups are revealed by the bands at 1727, 1387, 1248, and 1070 cm−1, associated with carboxyl, hydroxyl, epoxy, and carbonyl stretching frequency vibrations, respectively.21 The graphitic domains (C–C) at band 1624 cm−1 were observed for both GO and rGO. In case of rGO, the single peak at 1624 cm−1 indicated that the other oxygenated functional groups, such as carboxyl, hydroxyl, epoxy, and carbonyl, had nearly disappeared due to the reduction of GO and the formation of rGO. In all FTIR spectra, the peak at 2355 cm−1 indicated the presence of atmospheric CO2 (ref. 40) molecule. Raman spectroscopy results for CoMoO4, and GO and rGO are shown in Fig. 4A and B, respectively. The Raman spectrum of CoMoO4 (Fig. 4A) was measured over the range from 500 to 1200 cm−1. Raman vibrational modes were revealed at 939, 885, 814, and 699 cm−1 (Fig. 4A); the band at 939 cm−1 is associated with the symmetric stretching mode of the Mo–O bond, and the bands observed at 885 and 814 cm−1 correspond to the asymmetric stretching modes of oxygen in the O–Mo–O bond. The band located at 698 cm−1 was attributed to the symmetric stretching mode of the Co–O–Mo bond.30,41 The Raman spectra of GO and rGO were measured over the range of 1000–2500 cm−1 (Fig. 4B). Generally, the Raman spectrum of graphene-based materials is characterized by two main features: the G band and the D band; the former arises from first-order scattering of the E2g phonon of sp2-bonded carbon, while the latter is associated with vacancies, grain boundaries, and amorphous carbon species.42,43 The Raman spectra of the GO nanosheets show the presence of G and D bands at 1602 and 1356 cm−1, respectively. After hydrothermal reduction of GO into rGO, significant changes in the Raman spectra were observed: (i) the G band shifted toward a lower wave number, suggesting that re-graphitization had occurred, and (ii) the D band became more prominent due to an increase in the defect level from the reduction reaction.42 The specific surface area of the synthesized materials is evaluated using N2 adsorption–desorption isotherm (Fig. 5). The typical N2 adsorption–desorption isotherm of rGO is shown in Fig. 5A. The isotherm can be characterized as type IV, with hysteresis displayed over the relative pressure range from 0.4 to 0.9; this result indicates the availability of mesopores fractions.44 The obtained Brunauer–Emmett–Teller (BET) surface area (SBET) of hydrothermally synthesized rGO was 317.44 m2 g−1. Fig. 5B shows the N2 adsorption–desorption isotherm of CoMoO4. The measured BET surface area (SBET) of sonochemically synthesized CoMoO4 was 15.76 m2 g−1, which was mainly due to the porous structure of the CoMoO4 nanostructures.33
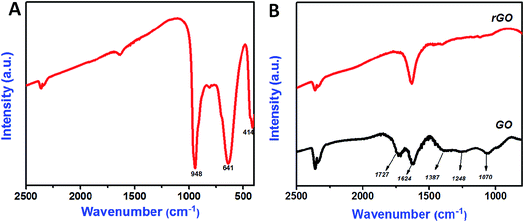 |
| | Fig. 3 FT-IR spectrum of CoMoO4 nanostructures (A), GO and rGO nanosheets (B). | |
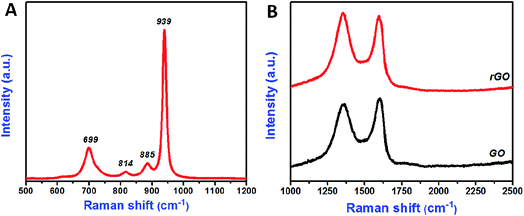 |
| | Fig. 4 Raman spectrum of CoMoO4 nanostructures (A), GO and rGO nanosheets (B). | |
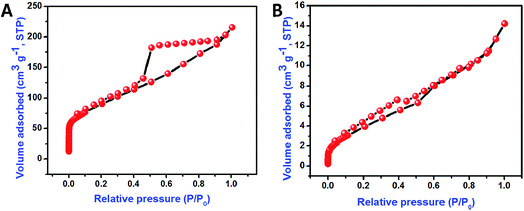 |
| | Fig. 5 Nitrogen adsorption–desorption isotherm of rGO nanosheets (A) and CoMoO4 nanostructures (B). | |
The electrochemical performance of the synthesized material was carried out using CV and GCD. Initially, the positive and negative electrodes were tested individually in 1.0 M NaOH using the three-electrode system. Fig. 6 shows the electrochemical performance of the rGO negative electrode; cyclic voltammograms were recorded over the potential range of −1 to 0 V, for a variable scan rate ranging from 5 to 100 mV s−1. Quasi-rectangular shapes were revealed, suggesting that the electrode had excellent electrochemical double-layer capacitance (Fig. 6A). The increase in the voltammetric current with the scan rate confirmed that the voltammetric current was directly proportional to the scan rate, an indicator of ideal capacitive behavior. The specific capacitance of the electrode materials was calculated using eqn (1):24
| | |
Specific capacitance (Csp) = [(∫IdV)(ν × ΔV × m)] F g−1
| (1) |
where
Csp is the specific capacitance (F g
−1), ∫
Id
V is the integral area of the CV curve,
ν is the scan rate (mV s
−1), Δ
V is the potential window (V), and
m is the mass of the active material (g).
Fig. 6B shows the calculated specific capacitances of the rGO electrode with respect to the current density. A specific capacitance of 168.8 F g
−1 was achieved at a scan rate of 5 mV s
−1; this value is comparable to that reported previously
45 and high compared with that obtained by Yan
et al.46 For a high scan rate of 100 mV s
−1, electrolyte ions can only access the outer surface of the electrode, which decreases both the active sites and the specific capacitance of the electrode material. For a low scan rate of 5 mV s
−1, the electrolyte ions can interact with both the inner and outer surfaces of the electrode material, resulting in a higher specific capacitance.
47
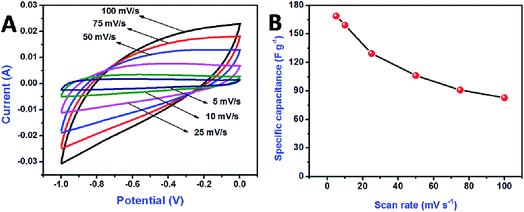 |
| | Fig. 6 Cyclic voltammetry curve for rGO electrode at the various scan rates (A), comparison of specific capacitances with respect to the scan rates (B). | |
Fig. 7 shows the electrochemical performance of CoMoO4 as a positive electrode. Cyclic voltammograms were recorded over the potential range of 0–0.5 V for scan rates varying from 5 to 100 mV s−1. The CV profile of CoMoO4 (Fig. 7A) shows the presence of a redox pair, indicating the pseudocapacitive behavior of the material. When the scan rate increased, the voltammetric current increased gradually, suggesting that the kinetics of the interfacial Faradaic redox reactions and the rates of electronic and ionic transport were sufficient for the given scan rates. Fig. 7B shows the calculated specific capacitance of the CoMoO4 electrode with respect to the current density. The maximum specific capacitance of 98.34 F g−1 was achieved for a scan rate of 5 mV s−1, which is comparable to that obtained in our previous studies.30,36
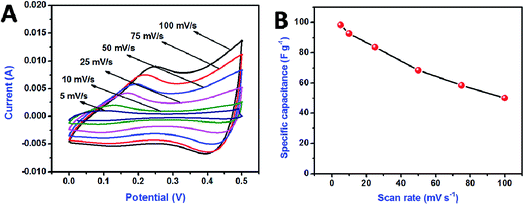 |
| | Fig. 7 Cyclic voltammetry curve for CoMoO4 electrode at the various scan rates (A), comparison of specific capacitances with respect to the scan rates (B). | |
An ASC was assembled using rGO as the negative electrode and CoMoO4 as the positive electrode, denoted as rGO‖CoMoO4 (Fig. 8A); the inset of Fig. 8A shows a photograph of the fabricated device. Fig. 8B shows the CV curve of both rGO and CoMoO4 for a scan rate of 10 mV s−1. The potential range for rGO was −1 to 0 V and that for CoMoO4 was 0 to 0.5 V versus Ag/AgCl. Therefore, the rGO‖CoMoO4 device was expected to achieve a maximum working voltage of 1.5 V. To attain the maximum device capacitance, the mass of the negative and positive electrodes should be balanced to equate the specific capacitance of the individual electrodes. Because the specific capacitances and the operational potential window of the positive and negative electrodes are different, the charge storage capacities of both electrodes were balanced by fine-tuning the mass loading between these electrodes. The CoMoO4 to rGO mass ratio was calculated using eqn (2) to achieve charge balance (q+ = q−),48
| | |
m−/m+ = (C+ × ΔV+)/(C− × ΔV−),
| (2) |
where
m− and
m+ represent the mass of the negative and positive electrodes,
C− and
C+ are the specific capacitance of the negative and positive electrodes, and, Δ
V−, Δ
V+ are the potential window of the negative and positive electrodes obtained using the three-electrode system, respectively. Subsequently, the ASC device was fabricated using rGO as the negative electrode and CoMoO
4 as the positive electrode in a 1.0 M NaOH electrolyte solution.
Fig. 8C shows the CV curves of the rGO‖CoMoO
4 ASC device measured over the voltage range of 0–1.5 V, for a scan rate of 100 mV s
−1. The results in
Fig. 8C confirm that the fabricated ASC device can operate with a potential window of 1.5 V, without instability. GCD analysis was also performed for operating voltages over the range of 0–1.5 V at current density of 0.5 mA cm
−2 (
Fig. 8D). The GCD curves at 0.5 mA cm
−2 stayed nearly symmetric behavior confirming the excellent capacitive behavior of the device over the entire voltage range.
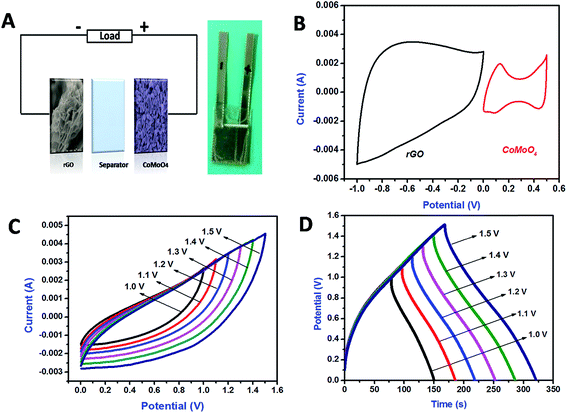 |
| | Fig. 8 Schematic representation of fabricated ASC (A), photograph of fabricated device (inset of A), cyclic voltammetry comparison of negative (rGO) and positive (CoMoO4) electrodes at the scan rate of 10 mV s−1 (B), cyclic voltammetry curve for fabricated ASC at the different voltage levels (100 mV s−1) (C), galvanostatic charge discharge curve for fabricated ASC at the different voltage levels (0.5 mA cm−2) (D). | |
Fig. 9A shows the CV curve of the ASC device, for a variable scan rate of 5–200 mV s−1. No significant change was observed in the nature of the CV curves, even for a scan rate of 200 mV s−1, which indicates the better rate capability of the fabricated asymmetric device. A maximum capacitance of 27.7 F g−1 was achieved for a scan rate of 5 mV s−1. Fig. 9B shows the variation in the specific capacitance as a function of the scan rate. The specific capacitance decreased as the scan rate increased; in this case, the electrolyte ions were not fully utilized by the electrode material due to the faster scan rates. Fig. 9C shows the GCD curve of the ASC collected at different current densities ranging from 0.5 to 1 mA cm−2. The fabricated device exhibited nearly symmetric charge–discharge profiles, suggesting good charge-storage capability. The specific capacitance (Csp), energy density (E), and power density (P) from charge–discharge analysis were calculated using eqn (3)–(5), respectively:23,49
| | |
Specific capacitance Csp = [(I × t)/m × V] F g−1,
| (3) |
| | |
Energy density E = (CspV2)/2 W h kg−1,
| (4) |
and
| | |
Power density P = E/t W kg−1
| (5) |
where
I is the discharge current (mA),
t is the discharge time (s),
m is the mass of the electroactive material (mg), and
V is the potential window (V).
Fig. 9D shows the variation in the specific capacitance with respect to the current density. A maximum specific capacitance of 26.16 F g
−1 was achieved for a current density of 0.5 mA cm
−2. The observed specific capacitance from the GCD analysis was well matched with the specific capacitance value obtained from CV measurements.
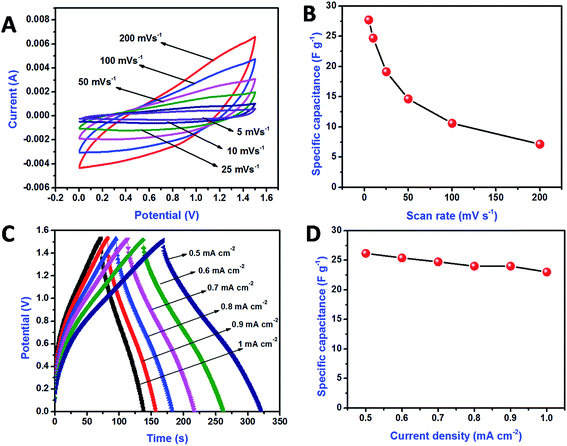 |
| | Fig. 9 Cyclic voltammetry curve for ASC at the various scan rates (A), comparison of specific capacitances with respect to the scan rates (B), charge discharge curve for ASC at the various current densities (C), comparison of specific capacitances with respect to the current densities (D). | |
The amount of energy stored and delivered by SC devices can be evaluated using the energy and power densities, which play an important role in the capacitive behavior of electrochemical devices. From the eqn (4) and (5),28 the calculated energy and power densities were 8.17 W h kg−1 and 187.5 W kg−1, respectively, for a current density of 0.5 mA cm−2. The cyclic stability of the fabricated rGO‖CoMoO4 ASC device was evaluated via GCD measurements for an operating voltage of 1.5 V and a current density of 1 mA cm−2. The ASC device exhibited superior cyclic performance, about 84.7% specific capacitance retention after 4000 cycles (Fig. 10); the decrease in capacitance was attributed to electrode aggregates in the electrolytes.50 Thus, our results demonstrated the excellent cyclic stability of the fabricated rGO‖CoMoO4 ASC device.
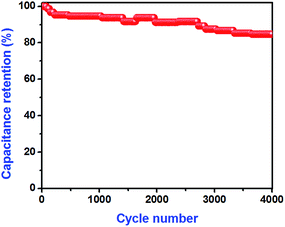 |
| | Fig. 10 Cyclic performance of fabricated ASC measured by the GCD measurement at the current density of 1.0 mA cm−2. | |
4. Conclusions
In summary, we demonstrated an ASC device composed of rGO negative electrode and CoMoO4 positive electrode. By integrating these positive and negative electrodes, the device achieved an operating voltage of 1.5 V, a maximum specific capacitance of 26.16 F g−1, an energy density of 8.17 W h kg−1, and a power density of 187.5 W kg−1, for a current density of 0.5 mA cm−2. Additionally, the fabricated device exhibited excellent cyclic stability (84.7% retention of its initial specific capacitance) over 4000 cycles of charge–discharge analysis at a current density of 1 mA cm−2. Collectively, our results show that the rGO‖CoMoO4 ASC device has great potential for application to energy storage devices.
Acknowledgements
This Research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2013R1A2A2A01068926).
References
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed.
- S. Roldan, Z. Gonzalez, C. Blanco, M. Granda, R. Menendez and R. Santamaria, Electrochim. Acta, 2011, 56, 3401–3405 CrossRef CAS PubMed.
- K. R. Prasad and N. Munichandraiah, Electrochem. Solid-State Lett., 2002, 5(12), A271–A274 CrossRef CAS PubMed.
- Y. Xu, I. Hennig, D. Freyberg, A. J. Strudwick, M. G. Schwab, T. Weitz and K. C. P. Cha, J. Power Sources, 2014, 248, 483–488 CrossRef CAS PubMed.
- B. Conway, Electrochemical supercapacitors, Kluwer Academic/Plenum, New York, 2000 Search PubMed.
- R. Költz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 CrossRef.
- F. Wang, S. Xiao, Y. Hou, C. Hu, L. Liu and Y. Wu, RSC Adv., 2013, 3, 13059–13084 RSC.
- Q. T. Qu, Y. Shi, L. L. Li, W. L. Guo, Y. P. Wu, H. P. Zhang, S. Y. Guan and R. Holze, Electrochem. Commun., 2009, 11, 1325–1328 CrossRef CAS PubMed.
- X. Hu, Y. Huai, Z. Lin, J. Suo and Z. A. Deng, J. Electrochem. Soc., 2007, 154, A1026–A1030 CrossRef CAS PubMed.
- D. W. Wang, H. T. Fang, F. Li, Z. G. Chen, Q. S. Zhong, G. Q. Lu and H. M. Cheng, Adv. Funct. Mater., 2008, 18, 3787–3793 CrossRef CAS.
- P. C. Chen, G. Shen, Y. Shi, H. Chen and C. Zhou, ACS Nano, 2010, 4, 4403–4411 CrossRef CAS PubMed.
- L. J. Xie, J. F. Wu, C. M. Chen, C. M. Zhang, L. Wan, J. L. Wang, Q. Q. Kong, C. X. Lv, K. X. Li and G. H. Sun, J. Power Sources, 2013, 242, 148–156 CrossRef CAS PubMed.
- B. X. Zou, Y. Liang, X. X. Liu, D. Diamond and K. T. Lau, J. Power Sources, 2011, 196, 4842–4848 CrossRef CAS PubMed.
- D. Salinas-Torres, J. M. Sieben, D. Lozano-Castelló, D. Cazorla-Amorós and E. Morallón, Electrochim. Acta, 2013, 89, 326–333 CrossRef CAS PubMed.
- Z. Niu, W. Zhou, J. Chen, G. Feng, H. Li, W. Ma, J. Li, H. Dong, Y. Ren, D. Zhao and S. Xie, Energy Environ. Sci., 2011, 4, 1440–1446 CAS.
- J. Cao, Y. Wang, Y. Zhou, J. H. Ouyang, D. Jia and L. Guo, J. Electroanal. Chem., 2013, 689, 201–206 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- K. Zhang, L. L. Zhang, X. S. Zhao and J. S. Wu, Chem. Mater., 2010, 22, 1392–1401 CrossRef CAS.
- J. Xia, F. Chen, J. Li and N. Tao, Nat. Nanotechnol., 2009, 4, 505–509 CrossRef CAS PubMed.
- F. Luan, G. Wang, Y. Ling, X. Lu, H. Wang, Y. Tong, X. X. Liu and Y. Li, Nanoscale, 2013, 5, 7984–7990 RSC.
- J. Yan, Z. Fan, W. Sun, G. Ning, T. Wei, Q. Zhang, R. Zhang, L. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632–2641 CrossRef CAS.
- H. Wang, C. M. B. Holt, Z. Li, X. Tan, B. S. Amirkhiz, Z. Xu, B. C. Olsen, T. Stephenson and D. Mitlin, Nano Res., 2012, 5(9), 605–617 CrossRef CAS.
- C. T. Hsu and C. C. Hu, J. Power Sources, 2013, 242, 662–671 CrossRef CAS PubMed.
- H. Peng, G. Ma, J. Mu, K. Sun and Z. Lei, J. Mater. Chem. A, 2014, 2, 10384–10388 CAS.
- H. Chen, J. Jiang, L. Zhang, T. Qi, D. Xia and H. Wan, J. Power Sources, 2014, 248, 28–36 CrossRef CAS PubMed.
- D. Guo, Y. Luo, X. Yu, Q. Lin and T. Wang, Nano Energy, 2014, 8, 174–182 CrossRef CAS PubMed.
- L. Q. Mai, F. Yang, Y. L. Zhao, X. Xu, L. Xu and Y. Z. Luo, Nat. Commun., 2011, 2, 381–385 CrossRef PubMed.
- G. K. Veerasubramani, K. Krishnamoorthy, S. Radhakrishnan and S. J. Kim, Mater. Chem. Phys., 2014, 147, 836–842 CrossRef CAS PubMed.
- M. C. Liu, L. B. Kong, X. J. Ma, C. Lu, X. M. Li, Y. C. Luo and L. Kang, New J. Chem., 2012, 36, 1713–1716 RSC.
- M. C. Liu, L. B. Kong, C. Lu, X. J. Ma, X. M. Li, Y. C. Luo and L. Kang, J. Mater. Chem. A, 2013, 1, 1380–1387 CAS.
- B. Senthilkumar, D. Meyrick, Y. S. Lee and R. K. Selvan, RSC Adv., 2013, 3, 16542–16548 RSC.
- K. Krishnamoorthy, K. Jeyasubramanian, M. Premanathan, G. Subbiah, H. S. Shin and S. J. Kim, Carbon, 2014, 72, 328–337 CrossRef CAS PubMed.
- G. K. Veerasubramani, K. Krishnamoorthy, S. Radhakrishnan, N. J. Kim and S. J. Kim, Int. J. Hydrogen Energy, 2014, 39, 5186–5193 CrossRef CAS PubMed.
- D. Y. Pan, S. Wang, B. Zhao, M. H. Wu, H. J. Zhang, Y. Wang and Z. Jiao, Chem. Mater., 2009, 21, 3136–3142 CrossRef CAS.
- K. Krishnamoorthy, M. Veerapandian, L. H. Zhang, K. Yun and S. J. Kim, J. Phys. Chem. C, 2012, 116, 17280–17287 CAS.
- J. W. Ward, J. Phys. Chem., 1968, 72(12), 4211–4223 CrossRef CAS.
- M. Sanati and A. Andersson, J. Mol. Catal., 1993, 81, 51–62 CrossRef CAS.
- A. P. de Moura, L. H. de Oliveira, P. F. S. Pereira, I. L. V. Rosa, M. S. Li, E. Longo and J. A. Varela, Adv. Chem. Eng. Sci., 2012, 2, 465–473 CrossRef.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22(35), 3906–3924 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. B. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- S. Yang, X. Feng, L. Wang, K. Tang, J. Maier and K. Müllen, Angew. Chem., Int. Ed., 2010, 49(28), 4795–4799 CrossRef CAS PubMed.
- D. Ma, Z. Wu and Z. Cao, J. Energy Chem., 2014, 23, 346–353 CrossRef CAS.
- J. Yan, T. Wei, B. Shao, F. Ma, Z. Fan, M. Zhang, C. Zheng, Y. Shang, W. Qian and F. Wei, Carbon, 2010, 48, 1731–1737 CrossRef CAS PubMed.
- Y. Wang, J. Chen, J. Cao, Y. Liu, Y. Zhou, J. H. Ouyang and D. Jia, J. Power Sources, 2014, 271, 269–277 CrossRef CAS PubMed.
- P. Ahuja, V. Sahu, S. K. Ujjain, R. K. Sharma and G. Singh, Electrochim. Acta, 2014, 146, 429–436 CrossRef CAS PubMed.
- Y. Hou, L. Chen, P. Liu, J. Kang, T. Fujita and M. Chen, J. Mater. Chem. A, 2014, 2, 10910–10916 CAS.
- K. Krishnamoorthy, G. K. Veerasubramani, S. Radhakrishnan and S. J. Kim, Chem. Eng. J., 2014, 251, 116–122 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 









