
An ab initio study of TiS3: a promising electrode material for rechargeable Li and Na ion batteries†
Jian Wua,
Da Wanga,
Hao Liub,
Woon-Ming Lauab and
Li-Min Liu*a
aBeijing Computational Science Research Center, Beijing 100084, China. E-mail: limin.liu@csrc.ac.cn
bChengdu Green Energy and Green Manufacturing Technology R&D Center, Chengdu Development Center of Science and Technology of CAEP, Chengdu, Sichuan 610207, China
First published on 9th February 2015
Abstract
Titanium trisulfide (TiS3) was recently reported to be highly promising as an electrode material for Li-ion batteries, due to its multielectron processes with high theoretical capacity. However, theoretical work on the performance and mechanism of Li adsorption in bulk and monolayer TiS3 is still lacking. The constraint of lithium resource also requires replacement by an abundant material such as Na. Using first principles calculations based on density functional theory, this study extensively investigates the electronic structure, adsorption and diffusion properties, capacity and plateaus of Li and Na atoms in bulk and monolayer TiS3. The results reveal that as the thickness of the TiS3 material decreased to a monolayer, a transition from an indirect band gap to a direct band gap was induced. Both the difference in charge density and the Bader charge analysis show that a significant charge transfer occurs from a Li or Na adatom to its neighboring sulfur atoms. Additionally, in bulk and monolayer TiS3, both Li and Na show two diffusion pathways with a low diffusion barrier, and one pathway can be further enhanced as the TiS3 changes from bulk to monolayer. Moreover, monolayer TiS3 shows a lower energy barrier for Na atoms, and there is also no problem associated with volume expansion in bulk TiS3. At high Li/Na concentrations, the Li/Na atoms can also diffuse easily, and one diffusion pathway is viable in bulk TiS3, which is effective for direct diffusion. All these properties are promising for the development of Li and Na batteries based on bulk and monolayer TiS3.
1. Introduction
Developing novel and efficient electrochemical storage device with high energy densities is vitally important for powering our future society. Among the promising candidates for next-generation high-energy rechargeable batteries, metal sulfides show great potential due to their high electronic conductivity, high theoretical density and low solubility in electrolytes.1–3 However, over a wide range of materials, there are only a few metal polysulfides that have a high sulfur content.In recent years, titanium polysulfides (TiSX) have captivated particular attention due to their potential applications as electrode materials, field effect transistors (NR-FET), hydrogen storage devices and in thermoelectric energy conversion.4–9 Among these polysulfides, crystalline titanium disulfide (TiS2) with layered structure exhibits excellent cyclability in both electrochemical cells with organic liquid electrolytes and all-solid-state cells with sulfide solid electrolytes.10–12 Crystalline titanium trisulfide (TiS3) is another promising candidate because its particles own an even higher theoretical capacity than that of TiS2 in liquid-type lithium cells. Lindic et al.13 found that the TiS3 cell exhibited a capacity of 350 mA h g−1 for the initial few cycles, and Hayashi et al.14 reported that amorphous TiS3 particles have a high reversible capacity of 400 mA h g−1 in all-solid-state batteries.
On the other hand, the constraint in the lithium resource leads to the requirement to replace lithium with other abundant elements, such as sodium (Na). Various Na based energy storage systems, including Na-ion batteries and Na-ion capacitors, have been widely explored.15–18 Unfortunately, although a high capacity is generally observed during the first charge in electrochemical reaction of TiS3 with Na, the capacity cannot be maintained during subsequent cycles owing to the structural decomposition of TiS3 at high temperature.19 Therefore, to utilize Na batteries in bulk TiS3 systems, the study seeks to understand the insertion, diffusion and volume expansion after Na absorbed in bulk TiS3.
It has been well known that as their thickness decreases to a few layers or even to a monolayer, the structure and electronic properties of certain layered materials change remarkably leading to new and unexpected properties and hence bringing in many important applications.20 More importantly, few-layer TiS3 nanoribbons made from exfoliation present remarkable field effect transistors (FET) characteristics, high photoresponse and fast switching rates.21,22 However, no experimental and theoretical study has been focused on the mechanism of Li/Na adsorption on monolayer TiS3. From the theoretical point of view, first-principles computational methods are playing an important role in understanding the electronic structure and the performances of electrode materials.23–28 This study intends to fill the gap.
In this work, for the first time first principles calculations were used to comparatively examine the Li/Na adsorption and diffusion in bulk and monolayer TiS3. The phase stability, electronic properties, capacity and plateaus of both bulk and monolayer TiS3 were carefully investigated. Meanwhile, the difference charge density and the Bader charge analysis were used to study the interaction between Li/Na adatom and its surrounding atoms. The energy barriers for Li and Na migration in bulk and monolayer TiS3 were also evaluated. These analyses would give us a solid foundation to better harness TiS3 as electrode materials for rechargeable Li and Na ion batteries.
2. Methods
The first-principles calculations were performed using DFT calculations as implemented in the Vienna ab initio simulation package (VASP).29,30 The exchange–correlation term has been described within the generalized gradient approximation (GGA) parameterized by Perdew–Burke–Ernzerhof (PBE) functional.31 A kinetic-energy cutoff for plane-wave expansion was set to 360 eV. A 2a × 3b × 1c supercell (where a, b and c are the lattice constants of bulk TiS3) with a three dimension period boundary condition for bulk, and a 2a × 3b slab with about 15 Å vacuum along the c direction for monolayer TiS3 were used to simulate the Li and Na adsorption and diffusion y. All the atoms in the unit cell were fully relaxed until the force on each atom was less than 0.01 eV Å−1. Electronic minimization was performed with a tolerance of 10−6 eV. The Brillouin-zone (BZ)32 sampling was carried out with a 6 × 8 × 1 Monkhorst-Pack grid for 2D sheets and a 6 × 8 × 3 Monkhorst-Pack grid for bulk. In order to avoid the disadvantages of severely underestimating band gaps by using the DFT-PBE calculation, the hybrid functional calculations with HSE06![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 were used, which is a better test to give relatively accurate band gaps for a wide range of materials. The van der Waals (vdW) interaction of bulk TiS3 is corrected by using the DFT-D2 approach.34 The stability of monolayer TiS3 is studied by calculating its phonon spectra based on the force constant approach35 with the package of Phonopy.
33 were used, which is a better test to give relatively accurate band gaps for a wide range of materials. The van der Waals (vdW) interaction of bulk TiS3 is corrected by using the DFT-D2 approach.34 The stability of monolayer TiS3 is studied by calculating its phonon spectra based on the force constant approach35 with the package of Phonopy.
To investigate the Li and Na diffusion kinetics, the study used the climbing image nudged elastic band (CI-NEB) method to seek saddle points and minimum energy pathway between the given initial and final positions. Hence, the energy barriers can be calculated. For the analyses, several images were employed between the fixed initial and final configurations. Each image was relaxed until the forces on atom were <0.01 eV Å−1.
3. Results and discussion
3.1 The structure and stability of TiS3
A full geometry optimization of both atomic coordinates and lattice parameters for both bulk and monolayer TiS3 were firstly performed. The optimized values are given in Table 1 together with experimental data. Fig. 1 shows that two of the three sulfur atoms in one formula unit are present as a single-bonded disulfide ion (S22−), and the third sulfur atom is formally sulfide (S2−). Bulk TiS3 consists of TiS3 layers attracted by a weak van der Waals interaction with a interlayer spacing of ∼3.2 Å. The calculated cell parameters and the interatomic Ti–S and disulfide (S22−) distances of bulk TiS3 agree well with experimental results.36 (Table 1) The atomic positions and cell parameters of monolayer TiS3 are almost the same as those in the bulk phase.| Bulk | Monolayer | Experiment36,37 | |
|---|---|---|---|
| a | 4.981 | 4.995 | 4.958 |
| b | 3.391 | 3.392 | 3.4006 |
| c | 8.910 | 8.778 | |
| Ti–S1 | 2.64 | 2.65 | 2.64 |
| Ti–S2 | 2.49 | 2.47 | 2.49 |
| Ti–S4 | 2.46 | 2.44 | 2.45 |
| S2–S3 | 2.04 | 2.01 | 2.04 |
| Band gap | 0.94 | 1.02 | 0.90 |
 | ||
| Fig. 1 (a) Structure of TiS3 (formula: Ti4+[S22−]S2−) extending over about eight unit cells: the blue and yellow balls represent the Ti and S atoms, respectively. S1 is sulfide (S2−), and S2 and S3 are disulfide (S22−). (b) and (c) are side views of bulk TiS3. | ||
For confirmation of the structure stability of monolayer TiS3, the full phonon dispersion spectrums of the structure along the high-symmetry directions in Brillouin zone were calculated. When a structure is thermodynamically stable, all its phonon frequencies on the k-points in the Brillouin zone should be positive.38–40 As shown in Fig. 2, almost no imaginary vibration frequency appears for the monolayer TiS3, implying that the monolayer TiS3 has high phonon stability. Thus, the monolayer TiS3 should be thermodynamically stable.
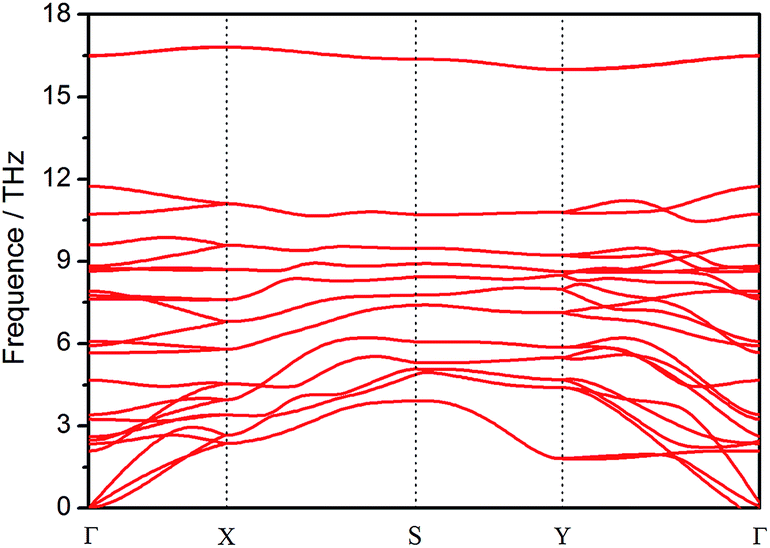 | ||
| Fig. 2 Calculated phonon branches of monolayer TiS3 along high-symmetry directions in the Brillouin zone. | ||
3.2 The electronic structure of TiS3
As it is known, the electronic conductivity of a material is extremely correlated with its electronic structures, especially for the ones whose energy band gap is around Fermi level. For comparison, the band structures and total density of states (DOS) of bulk and monolayer TiS3 are shown in Fig. 3(a) and (b), respectively. Based on the calculation with PBE, the bulk TiS3 is an indirect semiconductor with a band gap of 0.14 eV, which is about 0.76 eV smaller than the experimental value of Eg = 0.90 eV.37 It is well known that pure DFT usually underestimates the band gap of a semiconductor because of the self-interaction error. Therefore the hybrid functional HSE06 was used to examine the band gaps for both bulk and monolayer TiS3 as the hybrid functional can give more accurate band gap readings because of the introduction of the exact change. The calculated band gap of bulk TiS3 with HSE06 is 0.94 eV, which is in good agreement with the reported value above. | ||
| Fig. 3 Band structures and corresponding total density of states (DOS), Ef, shown by black dashed lines and dots, for (a) bulk and (b) monolayer TiS3, respectively. The Fermi energy is set to zero (By HSE06 calculations). | ||
Although the bulk TiS3 is an indirect semiconductor, interestingly, monolayer TiS3 becomes a direct one (see Fig. 3). The calculated band gap of monolayer TiS3 is 0.23 eV with PBE, while HSE06 gives a band gap of 1.02 eV, which is about 0.80 eV larger than that of bulk one. Such transition of band structure from indirect to direct has also been observed in other systems, such as MoS2, WS2, and MoSe2, during the exfoliation of the monolayer from the bulk materials.20,41 Besides larger surface area, monolayer TiS3 also processes special electronic properties, which may have important applications in battery and other fields.
3.3 TiS3 as Li/Na ion batteries anode
As discussed above, the calculated electronic structure of TiS3 shows that both bulk and monolayer TiS3 are semiconductors with a band gap of about 1 eV, which have potential applications in field effect transistors and optoelectronic switch. To put TiS3 into application as Li/Na ion batteries, we calculated the properties of Li/Na adsorption and diffusion in bulk and monolayer TiS3 as follows:Firstly, the Li/Na binding energy, Eb(Li/Na), between the Li/Na atom and the bulk and monolayer TiS3 are calculated by the following definition:
| Eb(Li/Na) = Etot(TiS3) + Etot(Li/Na) − Etot(TiS3–Li/Na), |
A schematic presentation of the atomic configuration of Li/Na adsorbed on monolayer TiS3 at H, T1 and T2 sites is shown in Fig. 4. As can be seen in Fig. 4, there are three different high symmetry sites for both bulk and monolayer TiS3 i.e. H, T1 and T2 sites. The Eb(Li/Na) of these three sites are hence presented in Table 2. Meanwhile, the equilibrium bond length between Li/Na and nearest neighbor S atoms for both bulk and monolayer TiS3, as well as the expansion of lattice parameters c after Li/Na absorbed in bulk TiS3 were also calculated.
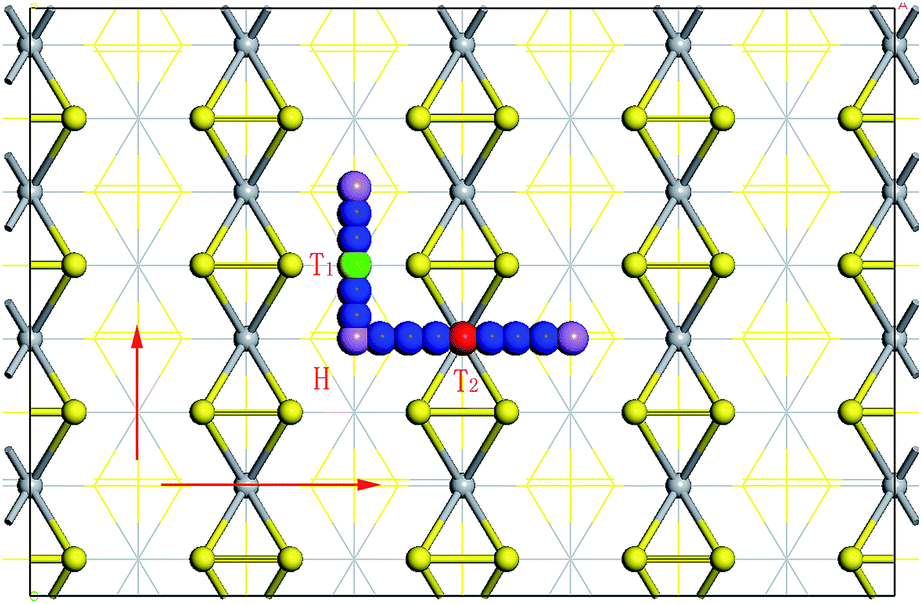 | ||
| Fig. 4 Top view of the atomic configuration of Li/Na adsorbed on monolayer TiS3 at H, T1 and T2 sites, respectively. The arrows show the diffusion pathway of Li/Na atoms between two neighboring H sites by passing through the T1 and T2 sites. | ||
| Monolayer | Bulk | |||
|---|---|---|---|---|
| Li | Na | Li | Na | |
| (H) E | 1.15 | 1.12 | 1.82 | 1.35 |
| Δc | 1.04% | 4.17% | ||
| dLi/Na–S | 2.44 | 2.81 | 2.52d, 2.53u | 2.68d, 2.67u |
| (T1) E | 0.69 | 0.78 | 1.71 | 1.18 |
| Δc | 1.77% | 6.56% | ||
| dLi/Na–S | 2.34 | 2.70 | 2.44d, 2.52u | 2.62d, 2.75u |
| (T2) E | 0.89 | 0.9 | 1.71 | 1.18 |
| Δc | 1.76% | 6.54% | ||
| dLi/Na–S | 2.46 | 2.81 | 2.52d, 2.44u | 2.75d, 2.62u |
For bulk TiS3, the six adjacent sulfur atoms form six coordination bonds with Li/Na atom in all three sites. The adatom on site H and T2 connect to the up layer with two bonds and the lower layer with the other four bonds, while the adatom on T1 site is in reverse to that on H and T2 site. The most stable site of adsorbed Li/Na atoms in bulk TiS3 is the H site, where the Li and Na binding energies are 1.82 and 1.35 eV, respectively. It should be noted that the Li/Na binding energies and the lattice expansion in c are the same in the T1 and T2 sites, because of the equivalent coordination bonds in the bulk. Compared with Li adsorption in bulk TiS3, the expansion of lattice parameters for Na adsorption is much higher, which will result in the structure deformation of TiS3 and then the capacitance fading. This is consistent with experimental result that bulk TiS3 is not suitable as electrodes for Na ion batteries.19
In the case of monolayer TiS3, the Eb(Li) for a Li adatom adsorbed on the H, T1 and T2 sites are 1.15, 0.69 and 0.89 eV, respectively (see Table 2). The most stable site of Li/Na adatom is the center of the four neighbor S atoms forming four coordination bonds in the same plane (H site). The equilibrium bond length of Li–S (dLi–S) is 2.44 Å, and the adsorbed Li atom is about 0.68 Å above the TiS3 layer. On the T2 site, Li is adsorbed on one Ti atom with a distance of 3.01 Å, forming four coordination bonds with slightly stretched dLi–S of 2.46 Å and greatly enlarged h of 1.33 Å. In the case of T1 site, Li is also adsorbed on one Ti atom with a distance of 5.96 Å but bonded with only two nearest neighbor S atoms with dLi–S of 2.34 Å, and the h is further enlarged (1.61 Å) compared with Li adatom on T2 site.
As with the Li adatom, study on Na adatom on the monolayer TiS3 surface at H, T1 and T2 sites was also carried out and the results are presented in Table 2. In general, the Na–S equilibrium bond lengths (dNa–S) for different position are elongated on average by 0.36 Å compared with dLi–S due to the larger atomic radius of Na.
To understand the bonding nature of Li/Na adsorbed in the bulk and monolayer TiS3, the study calculated the charge density difference, Δρ(r), as expressed in the following formula,
| Δρ(r) = ρLi/Na–TiS3(r) − ρLi/Na(r) − ρTiS3(r), |
Fig. 5 shows the charge density difference of Li/Na atoms located at the H site in bulk and monolayer TiS3. For the Li adsorbed at the H site on the monolayer TiS3, there is a net loss of electronic charge right above the Li, whereas there is a net gain of electronic charge in the intermediate region between Li and four adjacent sulfur atoms, indicating a significant charge transfer from the adsorbed Li to its nearest neighbor S atoms. The Bader charge analysis42,43 was performed to quantitatively estimate the amount of charge transfer between the adsorbed Li and the TiS3. The Bader charger state of a Li atom adsorbed at the H site on the monolayer TiS3 is +0.8678|e|, and the averaged Bader charge state of the four sulfur atoms next to the Li is −0.1488|e|. In the case of the Li located at the H site in the bulk TiS3, the Bader charge state of Li atom is +0.8602|e|, and the net gain of electronic charge for each adjacent sulfur atoms is −0.1298|e|. The total electronic transfer from Li and its nearest sulfur atoms in bulk TiS3 is −0.1836|e| higher than that in monolayer TiS3, leading to a higher binding energy between Li and S in bulk TiS3. Similar results can be found in Na adatom case. These results suggest that the interaction between the adsorbed Li/Na atom and its nearest neighbor sulfur atoms is predominantly ionic, and the valence electrons of the adsorbed Li/Na atoms are mainly transferred to the neighbor sulfur atoms.
 | ||
| Fig. 5 Isosurface (0.002 e Å), wathet blue for Δρ > 0 and dark blue for Δρ > 0 of the difference charge density Δρ for the most stable configuration of Li adsorbed at the H site in the (a), (b) monolayer and (c) bulk TiS3; and Na located at the H site in the (d), (e) monolayer and (f) bulk TiS3. | ||
As previously confirmed, Li and Na atoms tend to be adsorbed at the H site in both bulk and monolayer. Hence, Li/Na atoms could diffuse from an H site to a neighboring H site by passing through T1 or T2 site (see Fig. 4). Results of the investigation of diffusing barriers of Li/Na atoms through either site in both bulk and monolayer TiS3 are shown in Fig. 6 and Table 3.
 | ||
| Fig. 6 Energy barrier (∇E) for Li ((a) and (b)) and Na ((c) and (d)) atoms diffuse along H–T1–H and H–T2–H pathways in bulk and monolayer TiS3, respectively. | ||
| ∇E | Monolayer | Bulk | ||
|---|---|---|---|---|
| Li | Na | Li | Na | |
| H–T1–H | 0.44 | 0.32 | 0.31 | 0.24 |
| H–T2–H | 0.35 | 0.26 | 0.48 | 0.36 |
The diffusion barrier of Li/Na along the H–T2–H diffusion pathway decreases from 0.48/0.36 to 0.35/0.26 eV as the thickness decreased to a monolayer, respectively, which reveals that Li/Na can diffuse faster on monolayer TiS3 than in bulk TiS3. However, on monolayer TiS3, the calculated energy barriers (∇E) of Li/Na along the H–T1–H diffusion pathway are slightly higher compared with that in bulk TiS3. This can be attributed to the fact that the coordination bonds in monolayer TiS3 changes from four coordination bonds (H site) to two coordination bonds (T1 site) and then back to four coordination bonds (H site) during the Li/Na diffusion process. The changes of coordination bonds result in a larger energy barriers. On the other hand, no coordination bonds change in the case of bulk TiS3. These results indicate that the Li/Na mobility can be further enhanced along the H–T2–H pathway by decreasing the thickness of TiS3 to a monolayer. In addition, the energy barrier of Na is smaller than that of Li regardless of the thickness and the pathways, which can be attributed to the larger Na–S chemical bonds than Li–S and thus less constraint.
To further understand the Li/Na diffusion in the case of high Li/Na concentrations, we have investigated the energy barriers for Li/Na atoms diffusion along H–T1–H and H–T2–H pathways in bulk and monolayer Li/NaTiS3 systems, respectively. A schematic of the diffusion pathways are shown in Fig. 7 and the calculated diffusing barriers are summarized in Fig. 8 and Table. S1.† It is shown that at high Li/Na concentration, energy barriers for Li/Na atoms diffuse along H–T1–H and H–T2–H pathways in bulk and monolayer TiS3 are about 0.2 eV higher than that at low concentration cases. Moreover, it is interesting to find that the diffusion barrier of Na along the H–T2–H diffusion pathway in bulk TiS3 is closed due to the very high diffusion barrier. The increased diffusion barrier can be attributed to many factors, such as the increased electrostatic repulsion between Li/Na atoms, the volume expansion and lattice distortion at high Li/Na concentration. Furthermore, the energy barriers of Li are smaller than that of Na in both bulk and monolayer TiS3 due to its smaller radius. Our results suggest that the Li/Na diffuses slowly as the increase of Li/Na concentration.
 | ||
| Fig. 7 Schematic of the diffusion pathway of Li/Na atoms between two neighboring H sites by passing through the T1 and T2 sites in bulk and monolayer TiS3 with Li/Na atoms absorbed the whole H sites except an unoccupied H site, respectively. | ||
 | ||
| Fig. 8 Energy barrier (∇E) for Li ((a) and (b)) and Na ((c) and (d)) atoms diffuse along H–T1–H and H–T2–H pathways in bulk and monolayer TiS3 with Li/Na atoms absorbed the whole H sites except an unoccupied H site, respectively. | ||
In order to know the capacity of Li/Na in bulk and monolayer TiS3, we calculated the maximum Li/Na concentration. Based on our calculations, we found that bulk TiS3 can accommodate 2 Li/Na, occupying all the H, T1 and T2 sites. As a result, 339 and 282 mA h g−1 for Li and Na storage were obtained in bulk TiS3. However, in the case of monolayer TiS3, although the theoretical capacity of 488 and 377 mA h g−1 for Li and Na storage if all three adsorption sites are occupied, only 1.5 Li/Na can be adsorbed due to the serious structure deformation and weaker bonding strength of Li/Na at higher Li/Na concentrations. Consequently, the capacities of monolayer TiS3 are 260 and 225 mA h g−1 for Li and Na, respectively.
We further calculate the volume changes for the insertion of 1, 1.5 and 2 Li/Na atoms in bulk TiS3, respectively (Fig. S1†) It shows that the volume of bulk TiS3 expands with increasing Li/Na concentrations, and Na shows higher volume expansion than Li. In the case of 2 Li/Na atoms in bulk TiS3, volumes expanse by about 33 and 57% for Li/Na, respectively. Meanwhile, the bond length of two single-bonded disulfide ions (S22−) is broken from 2.04 Å to 3.06 Å and 3.19 Å for Li/Na, respectively, in agreement with the experimental result (Fig. S2†).13
To explore more information on the TiS3 as electrode material for Li and Na batteries, we calculated the voltage profile, the electrode potential V respect to Li/Li+ and Na/Na+ are expressed as follows,44,45
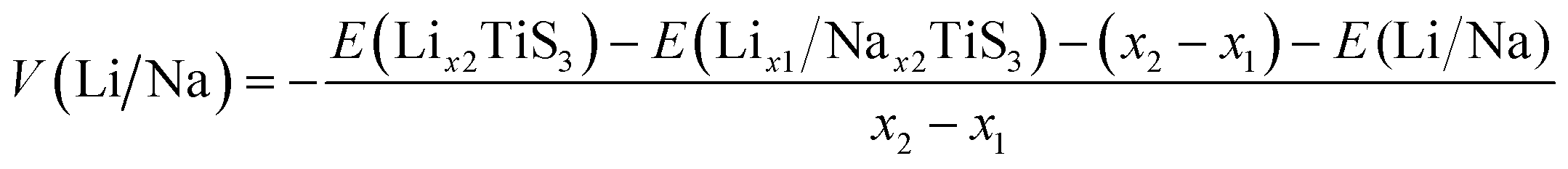
 | ||
| Fig. 9 Electrode potential of Li/Na-intercalated (a) bulk and (b) monolayer TiS3 against Li/Li+ and Na/Na+, respectively. | ||
From the above discussions, monolayer TiS3 shows a potential application in practical lithium batteries. However, because of the lower voltage of monolayer LixTiS3 compared to bulk, the weaker constraint of the surface S are more likely to react with Li or Na adatoms. Thus, it is essential for us to confirm the thermodynamic stability of monolayer LixTiS3 or NaxTiS3 within the charge–discharge process. The safety problem in metal sulfides often arises from the interaction of the highly charged S2− with the gradually filled Li/Na atoms. Recently, Lemmon and co-workers47 reported that MoS2 was reduced to metallic Mo and Li2S at 0.01 V (vs. Li+/Li). Fang et al.48 suggested that the lithium storage mechanism of MoS2 could be expressed as MoS2 + xLi+ + xe → LixMoS2 (above 1.00 V), and then followed by LixMoS2 + (4 − x)Li+ + (4 − x)e− → Mo + Li2S (0.01 V). Moreover, similar reaction mechanism was also found in the SnS2 system.49 In TiS3 system, when a large amount of S22− is present, these particularly unstable reduction states have the potential to further reduce by liberating S to form the energetic stable Li2S, yielding the great loss of active mass of the electrode and consequently a much limited cycle life of the battery. The intrinsic thermal stability of TiS3 can be understood by the following decomposition reaction
| AxTi12S36 + 2A = AxTi12S35 + A2S ΔE = E0(AxTi12S35) + E*(A2S) − E0(AxTi12S36) − 2E(A) |
4. Conclusions
In this paper, first-principle calculations have been used to study the electronic properties of bulk and monolayer TiS3 and its characteristics as electrode materials in rechargeable Li and Na ion batteries. The investigation of the electronic structure reveals that both bulk and monolayer TiS3 are semiconductors with a band gap of about 1 eV, but as the thickness of the TiS3 material decreases to a monolayer, a transition from an indirect band gap to a direct band gap occurs. The investigation on the adsorption and diffusion properties of Li and Na atoms in bulk TiS3 reveals that the energy diffusion barriers for both are low, and after absorption of Li and Na in bulk TiS3, Na shows higher expansion lattice parameters than Li. However, the volume expansion problem can be neglected in monolayer structures. Furthermore, the Li/Na mobility on monolayer TiS3 is improved along the H–T2–H pathway. The bulk TiS3 can accommodate 2 Li/Na, slightly higher than that of monolayer TiS3 of 1.5 Li/Na, corresponding to 339 and 282 mA h g−1 for Li and Na storage, respectively. Our calculated plateaus for Li in bulk TiS3 are in agreement with experiment. The plateaus for Li and Na in bulk TiS3 are higher than that in monolayer TiS3. Moreover, the plateaus against Li/Li+ are higher than that of against Na/Na+. In conclusion, our work confirms the following: (1). Bulk and monolayer TiS3 are semiconductors with low diffusion barriers and are therefore suitable materials for ion batteries. (2). Na is a viable replacement material for Li in TiS3, where Na shows better diffusion performance than Li. (3). The monolayer TiS3 shows improved Na ion batteries performance with smaller structure expansion compared with their bulk counterpart. (4). In bulk TiS3 with high Li/Na concentration, there is only one diffusion pathway, which is useful for direct diffusion.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 51222212), the CAEP foundation (Grant no. 2012B0302052 and 2013A030214), the financial support of Laboratory of Precision Manufacturing Technology, CAEP (ZZ13007), the MOST of China (973 Project, Grant no. 2011CB922200).Notes and references
- H. S. Kim, T. S. Arthur, G. D. Allred, J. Zajicek, J. G. Newman, A. E. Rodnyansky, A. G. Oliver, W. C. Boggess and J. Muldoon, Nat. Commun., 2011, 2, 427 CrossRef PubMed.
- M. S. Whittingham, Science, 1976, 192, 1126 CAS.
- L. Yu, L. Zhang, H. B. Wu and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 3711 CrossRef CAS PubMed.
- A. Sakuda, N. Taguchi, T. Takeuchi, H. Kobayashi, H. Sakaebe, K. Tatsumi and Z. Ogumi, Electrochem. Commun., 2013, 31, 71 CrossRef CAS PubMed.
- J. J. Ma, X. Y Liu, X. J Cao, S. H. Feng and M. E. Fleet, Eur. J. Inorg. Chem., 2006, 2006, 519 CrossRef.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 6139 CrossRef.
- Z. Y. Zeng, C. L. Tan, X. Huang, S. Y. Bao and H. Zhang, Energy Environ. Sci., 2014, 7, 797 CAS.
- C. T. Li, C. P. Lee, Y. Y. Li, M. H. Yeh and K. C. Ho, J. Mater. Chem. A, 2013, 1, 14888 CAS.
- N. A. Kyeremateng, N. Plylahan, A. C. S. dos Santos, L. V. Taveira, L. F. P. Dick and T. Djenizian, Chem. Commun., 2013, 49, 4205 RSC.
- M. S. Whittingham, Prog. Solid State Chem., 1978, 12, 41 CrossRef CAS.
- J. E. Trevey, C. R. Stoldt and S. H. Lee, J. Electrochem. Soc., 2011, 158, A1282 CrossRef CAS PubMed.
- C. M. Julien, Mater. Sci. Eng., R, 2003, 40, 47 CrossRef.
- M. H. Lindic, H. Martinez, A. Benayad, B. Pecquenard, P. Vinatier, A. Levasseur and D. Gonbeau, Solid State Ionics, 2005, 176, 1529 CrossRef CAS PubMed.
- A. Hayashi, T. Matsuyama, A. Sakuda and M. Tatsumisago, Chem. Lett., 2012, 41, 886 CrossRef CAS.
- Y. J. Kim, Y. Kim, A. Choi, S. Woo, D. Mok, N. S. Choi, Y. S. Jung, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2014, 26, 4139 CrossRef CAS PubMed.
- S. Y. Hong, Y. Kim, Y. Park, A. Choi, N. S. Choi and K. T. Lee, Energy Environ. Sci., 2013, 6, 2067 CAS.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Rojo, Energy Environ. Sci., 2012, 5, 5884 CAS.
- V. V. Kulish, O. I. Malyi, M. F. Ng, Z. Chen, S. Manzhos and P. Wu, Phys. Chem. Chem. Phys., 2014, 16, 4260 RSC.
- Z. Margherita, J. L. Shaw and G. J. Tennenhouse, J. Electrochem. Soc., 1981, 128, 1647 CrossRef PubMed.
- M. S. Xu, T. Liang, M. M. Shi and H. Z. Chen, Chem. Rev., 2013, 113, 3766 CrossRef CAS PubMed.
- J. O. Island, M. Buscema, M. Barawi, J. M. Clamagirand, J. R. Ares, C. Sánchez, I. J. Ferrer, G. A. Steele, H. S. J. van der Zant and A. Castellanos-Gomez, Adv. Opt. Mater., 2014, 2, 641 CrossRef CAS.
- I. J. Ferrer, J. R. Ares, J. M. Clamagirand, M. Barawi and C. Sánchez, Thin Solid Films, 2013, 535, 398 CrossRef CAS PubMed.
- J. Carrasco, J. Phys. Chem. C, 2014, 118, 19599 CAS.
- Y. Z. Chen, F. Peng, Y. Yan, Z. W. Wang, C. L. Sun and Y. M. Ma, J. Phys. Chem. C, 2013, 117, 13879 CAS.
- G. C. Yang, Y. C. Wang and Y. M. Ma, J. Phys. Chem. Lett., 2014, 5, 2516 CrossRef CAS.
- J. Zhou, Q. Wang, Q. Sun and P. Jena, J. Phys. Chem. C, 2011, 115, 6136 CAS.
- M. Kan, J. Y. Wang, X. W. Li, S. H. Zhang, Y. W. Li, Y. Kawazoe, Q. Sun and P. Jena, J. Phys. Chem. C, 2014, 118, 1515 CAS.
- D. Wang, L.-M. Liu, S.-J. Zhao, B.-H. Li, H. Liu and X.-F. Lang, Phys. Chem. Chem. Phys., 2013, 15, 9075 RSC.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188 CrossRef.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- X. Wu, M. C. Vargas, S. Nayak, V. Lotrich and G. Scoles, J. Chem. Phys., 2001, 115, 8748 CrossRef CAS PubMed.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B, 2008, 78, 134106 CrossRef.
- M. E. Fleet, S. L. Harmer, X. Liu and H. W. Nesbitt, Surf. Sci., 2005, 584, 133 CrossRef CAS PubMed.
- H. G. Grimeiss, A. Rabenau, H. Hahn and P. Ness, Z. fur Elektrochemie, 1961, 65, 776 Search PubMed.
- Q. Li, D. Zhou, W. T. Zheng, Y. M. Ma and C. F. Chen, Phys. Rev. Lett., 2013, 110, 136403 CrossRef.
- W. J. Yin, Y. E. Xie, L. M. Liu, R. Z. Wang, X. L. Wei, L. Lau, J. X. Zhong and Y. P. Chen, J. Mater. Chem. A, 2013, 1, 5341 CAS.
- H. Zhang, L. M. Liu and W. M. Lau, J. Mater. Chem. A, 2013, 1, 10821 CAS.
- S. Z. Butler, S. M. Hollen, L. Y. Cao, Y. Cui, J. A. Gupta, H. R. Gutiérrez, T. F. Heinz, S. S. Hong, J. Huang, A. F. Ismach, E. Johnston-Halperin, M. Kuno, V. V. Plashnitsa, R. D. Robinson, R. S. Ruoff, S. Salahuddin, J. Shan, L. Shi, M. G. Spencer, M. Terrones, W. Windl and J. E. Goldberger, ACS Nano, 2013, 7, 2898 CrossRef CAS PubMed.
- R. F. W. Bader, in Encyclopedia of Computational Chemistry, John Wiley & Sons, Ltd, 2002 Search PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354 CrossRef PubMed.
- M. K. Aydinol, A. F. Kohan, G. Ceder, K. Cho and J. Joannopoulos, Phys. Rev. B, 1997, 56, 1354 CrossRef CAS.
- M. K. Aydinol, A. F. Kohan and G. Ceder, J. Power Sources, 1997, 68, 664 CrossRef CAS.
- M. Mortazavi, C. Wang, J. K. Deng, V. B. Shenoy and N. V. Medhekar, J. Power Sources, 2014, 268, 279 CrossRef CAS PubMed.
- J. Xiao, D. Choi, L. Cosimbescu, P. Koech, J. Liu and J. P. Lemmon, Chem. Mater., 2010, 22, 4522 CrossRef CAS.
- X. P. Fang, X. W. Guo, Y. Mao, C. X. Hua, L. Y. Shen, Y. S. Hu, Z. X. Wang, F. Wu and L. Q. Chen, Chem.–Asian J., 2012, 7, 1013 CrossRef CAS PubMed.
- I. Lefebvre-Devos, J. Olivier-Fourcade, J. C. Jumas and P. Lavela, Phys. Rev. B, 2000, 61, 3110 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra15055d |
| This journal is © The Royal Society of Chemistry 2015 |