
Sonication-induced effects on carbon nanofibres in composite materials
Abstract
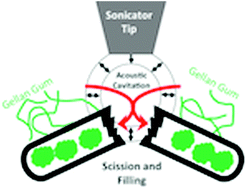
The preparation and characterization of carbon nanofibre–gellan gum composite materials is presented. Electron microscopy analysis reveals that nanofibres are affected by sonolysis, i.e. fibre length reduces, while filling occurs. Spectroscopic analysis suggests that the nanofibres are modified during the preparation of the dispersions. It is shown that despite these effects, composite materials prepared using a short period of sonolysis (4 min) exhibit robust conductivity, strain at failure and Young's modulus values of 35 ± 2 S cm−1, 20 ± 1% and 1.3 ± 0.3 MPa, respectively.
 Please wait while we load your content...
Please wait while we load your content...

