
LiFePO4 nanoparticles growth with preferential (010) face modulated by Tween-80†
Abstract
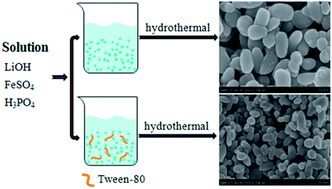
Small grain size combined with large I(020)/I(111) ratio have been proven to be an effective strategy to improve the electrochemical properties of LiFePO4 material due to increased Li-ion diffusion. In view of this, we use Tween-80 as the surfactant in hydrothermal synthesis to modulate both crystal size and orientation of LiFePO4. It is indicated that the LiFePO4 particles synthesized via the Tween-80 modified hydrothermal method exhibited a small mean diameter of 100 nm and a large I(020)/I(111) ratio of 1.19. Whereas in the surfactant-free hydrothermal system, the obtained LiFePO4 particles exhibit a relatively large mean diameter of 200 nm and a low I(020)/I(111) ratio of 0.84. LiFePO4 particles with small grain size and a large I(020)/I(111) ratio can be easily brought into contact with electrolyte, facilitating electric and Li-ion diffusion. After being coated with conductive carbon, the synthesized LiFePO4/C nanoparticles perform a high Li-ion diffusion coefficient of 1.79 × 10−13 cm2 s−1. It presents a large reversible capacity of 166.5 mA h g−1 at 0.1 C and even a high rate capacity of 119.6 mA h g−1 at 20 C.
 Please wait while we load your content...
Please wait while we load your content...

