
Amine functionalized tetraphenylethylene: a novel aggregation-induced emission based fluorescent chemodosimeter for nitrite and nitrate ions†
Amrita Chatterjee*a,
Dipratn G. Khandarea,
Praveen Sainia,
Anjan Chattopadhyaya,
Mahesh S. Majikbc and
Mainak Banerjee*a
aDepartment of Chemistry, BITS, Pilani – K. K. Birla Goa Campus, NH 17 B Bypass Road, Zuarinagar, Goa 403726, India. E-mail: amrita@goa.bits-pilani.ac.in; mainak@goa.bits-pilani.ac.in; Fax: +91-832-2557-033; Tel: +91-832-2580-347 (M. B.) Tel: +91-832-2580-320 (A. C.)
bBio-organic Chemistry Laboratory, CSIR-National Institute of Oceanography, Dona-Paula, Goa 403 004, India
cDepartment of Chemistry, Goa University, Taleigao Plateau, Goa 403 206, India
First published on 24th March 2015
Abstract
A novel AIE-based fluorescent probe for the detection of trace amounts of nitrite and nitrate ions in water has been developed. The probe, a monoamine of tetraphenylethylene, spontaneously detects nitrites (or nitrates) by a fluorescence “turn-off” method via diazotization followed by formation of a non fluorescent TPE-azodye. The salient features of this method are high sensitivity and selectivity, cost effective synthesis, fast detection process and low detection limit.
The dramatic increase in the concentration of nitrite and nitrate ions in groundwater, rivers and lakes from chemical fertilizers, livestock waste, etc. is a growing threat to public health and the environment.1 Nitrite is well-known to have key physiological roles in blood flow regulation, signaling and hypoxic nitric oxide homeostasis.2 It reacts with dietary components in the stomach to generate carcinogenic nitrosamine.3 Another latent hazard with nitrite ions is their capability to change oxyhemoglobin into methemoglobin when present in the bloodstream, thereby interfering with oxygen transport in the blood.4 Nitrate, though less toxic, is converted to nitrite by microbial reduction under physiological conditions.4 High concentrations of nitrate and nitrite ions in drinking water results in a number of medical issues, such as premature birth, intrauterine growth restriction and birth defects of the central nervous system.4 Therefore, the monitoring of nitrite (and nitrate) levels in drinking water and food stuffs is of great importance.
To date, a large number of techniques have been developed for the detection of nitrite ions, based on organic chromophores,5 electrochemical detection,6 ion chromatography,7 and others.8 Many of these available methods require the use of sophisticated instruments and thus are not cost-effective. In addition, some of these are not sufficiently sensitive or selective for the determination of trace amounts of nitrite ions, and hence cannot be used for real-time applications. Recently, a few sensitive chromogenic sensors have been reported for nitrite ions.9 However, over the last decade, fluorimetric probes have brought about a revolution in sensing technology because of their operational simplicity and cost-effectiveness, in addition to high sensitivity and selectivity.10 Surprisingly, molecular probes for nitrite ions based on the fluorimetric technique are rare.11 Therefore, it is highly desirable to develop new and sensitive methods for the determination of trace levels of nitrite ions in water and other substances. However, conventional organic fluorophores have a major inadequacy, despite several advantages. Although they are highly emissive in dilute solutions, the aggregation-caused quenching effect (ACQ) in the condensed phase restricts their real world applications.12 Recently, the advancement of luminogen molecules, which emit more effectively in the aggregated form than in the solution due to the aggregation-induced emission (AIE) mechanism, has stimulated great research interest for various applications.13–17 Using this novel phenomenon, a great variety of AIE active molecules have been designed for cell imaging,16a,d,e,17i optical devices,17h,j electroluminescent materials,15f,16e chemo/biosensors,13,15,17a,b,d,g,k etc. Tetraphenylethylene (TPE), due to its easy synthesis and simple functionalization strategies, is one of the most studied luminophores and the demonstration of its practical applications for the detection of analytes15 and for other purposes16 has been investigated in recent times. However, to the best of our knowledge, the aggregation-induced emission property has not been exploited to develop a sensor for the detection of nitrite ions to date. In this regard, we envisioned that a TPE-amine may serve as an efficient sensor for nitrite ions by making use of a simple “diazotization” reaction. As a part of our continued effort for the development of fluorescent sensors for biologically/environmentally important analytes,11a,15l,m,18 we report, herein, an AIE-active TPE-based fluorescent chemodosimeter for selective and sensitive detection of nitrite (and nitrate) ions in contaminated water.
For this sensing purpose, probe 1 (Fig. 1) was synthesized using a McMurry cross-coupling reaction, from benzophenone and 4-aminobenzophenone in a single-step, adopting a reported procedure (see ESI† for details).19 The probe readily undergoes “diazotization” by the reaction with nitrite ions in acidic water at 0–5 °C. Upon addition of an alkaline β-naphthol solution, a TPE-azodye congener (2) is formed, which is non-fluorescent in either the solid state or in solution (Fig. 1).
 | ||
| Fig. 1 The sensing mechanism for the detection of nitrite ions. | ||
As anticipated, probe 1 was non-emissive in THF solution, and since its solubility is very poor in water the addition of a large volume of water or mild acid (viz. 0.1 N HCl) preserves an intense fluorescence response under identical conditions, manifesting the AIE behaviour. As the diazotization process requires acidic conditions, the solvent dependent AIE behaviour of the probe was examined in THF and aqueous HCl. The fluorescence signal from the solution of probe 1 started to appear once the volume of THF in 0.1 N aqueous HCl (pH 1) was reduced to only 10% and approached a maximum at and below 3% THF in 0.1 N aqueous HCl (Fig. 2). As a preliminary study, we checked the ability of probe 1 to detect nitrite ions in 20% THF in water at pH 1. We expected a turn-on type fluorescence response from the probe in the presence of nitrite ions due to the anticipated poor solubility of the resultant azo-dye (2) in the same solvent system. To our surprise, a non-fluorescent solution of 1 did not show any fluorescence response even with the addition of one equiv. of nitrite. Moreover, the solid dye settled down to the bottom, and as an added surprise, the dye was found to be non-fluorescent in the solid state as well. This unexpected outcome prompted us to carry out a thorough investigation of the theoretical aspects of the electronic ground and excited states of both 1 and 2. We also established the optimum conditions for the diazotization reaction by measuring the fluorescence intensity of various solutions at different pH values, containing 1 equiv. of nitrite ions at 0–5 °C. A pH range of 1–7 was studied and pH 1 was found to be most suitable in terms of the time required for completion of the reaction (Fig. S1 of ESI†). The rate of diazotization becomes slower and slower up to pH 4.
 | ||
| Fig. 2 Plot of fluorescence intensity of probe 1 (30 μM) against various proportions of a 0.1 N HCl–THF mixture (λex 345 nm, λem 447 nm). | ||
As expected, the probe does not work at or near neutral pH. Low temperature, a requirement of conventional diazotization reactions, was maintained for all fluorimetric studies to avoid any side reactions.
To understand the ground and excited state behaviours, quantum mechanical studies were carried out at the density functional theory (DFT) and time-dependent density functional theory (TD-DFT) levels using the Gaussian 09 program (see ESI† for details). The optimized structures showed that the ethylenic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (1.37 Å) of probe 1 almost became a single bond (1.48 Å) in the photo-excited singlet state, while the adjacent C–C single bond lengths were found to decrease by 0.02 to 0.05 Å. The S0–S1 transition arises due to the HOMO → LUMO excitation and this was characterized by a movement of the π-cloud from the aniline moiety towards the central ethylenic part (Fig. S3 of ESI†). However, the non-coplanarity of the phenyl rings and the central C–C bond are likely to hinder the extensive π-delocalization in both the ground and excited states. A better co-planarity, resulting in improved delocalization, can be expected in the aggregated form and might be the reason behind the fluorescent nature of TPE-amine 1 in this state. However, the azo dye 2 was found to be non-fluorescent in nature, both in the solution and in the solid state. A major fraction of the π-cloud from its HOMO was found to be shifted away from the TPE-moiety and resided close to the azo bond and naphthalene moiety in the LUMO. The π electronic cloud was found to disappear from the three phenyl rings and the central ethylene part of the TPE moiety on HOMO → LUMO excitation (Fig. 3). The phenyl ring of the TPE moiety connected to the electron withdrawing azo bond and the naphthalene moiety situated on the other side of this bond are co-planar, and create a channel for shifting the π-cloud from TPE to this part during the S0–S1 transition. The disappearance of the π electron cloud on the major portion of the TPE moiety during the HOMO → LUMO excitation is probably related to the observed nonfluorescent nature of the azo-dye (2). This photo-induced intramolecular charge transfer process is subsequently followed by a non-radiative decay path from the charge separated excited state. The energy gap between the excited state at its optimized geometry and the ground state at this geometry was found to be negligibly small, which indicates a possibility of strong vibronic coupling between the states, leading to a non-radiative decay of the singlet excited state (see ESI†). Moreover, neither is the aggregated state expected to have better π-delocalization (even if it becomes completely planar) as the electron withdrawing part of the dye will still inhibit any π-electronic cloud over the TPE part.
C bond (1.37 Å) of probe 1 almost became a single bond (1.48 Å) in the photo-excited singlet state, while the adjacent C–C single bond lengths were found to decrease by 0.02 to 0.05 Å. The S0–S1 transition arises due to the HOMO → LUMO excitation and this was characterized by a movement of the π-cloud from the aniline moiety towards the central ethylenic part (Fig. S3 of ESI†). However, the non-coplanarity of the phenyl rings and the central C–C bond are likely to hinder the extensive π-delocalization in both the ground and excited states. A better co-planarity, resulting in improved delocalization, can be expected in the aggregated form and might be the reason behind the fluorescent nature of TPE-amine 1 in this state. However, the azo dye 2 was found to be non-fluorescent in nature, both in the solution and in the solid state. A major fraction of the π-cloud from its HOMO was found to be shifted away from the TPE-moiety and resided close to the azo bond and naphthalene moiety in the LUMO. The π electronic cloud was found to disappear from the three phenyl rings and the central ethylene part of the TPE moiety on HOMO → LUMO excitation (Fig. 3). The phenyl ring of the TPE moiety connected to the electron withdrawing azo bond and the naphthalene moiety situated on the other side of this bond are co-planar, and create a channel for shifting the π-cloud from TPE to this part during the S0–S1 transition. The disappearance of the π electron cloud on the major portion of the TPE moiety during the HOMO → LUMO excitation is probably related to the observed nonfluorescent nature of the azo-dye (2). This photo-induced intramolecular charge transfer process is subsequently followed by a non-radiative decay path from the charge separated excited state. The energy gap between the excited state at its optimized geometry and the ground state at this geometry was found to be negligibly small, which indicates a possibility of strong vibronic coupling between the states, leading to a non-radiative decay of the singlet excited state (see ESI†). Moreover, neither is the aggregated state expected to have better π-delocalization (even if it becomes completely planar) as the electron withdrawing part of the dye will still inhibit any π-electronic cloud over the TPE part.
 | ||
| Fig. 3 Frontier molecular orbital diagrams of HOMO and LUMO of azo-dye 2 at optimized ground state geometry. | ||
For the sensing study, we selected 3% THF in 0.1 N HCl as the most suitable solvent system in which probe 1 fluoresces with the highest intensity. In a typical experiment, the fluorescence response of the highly fluorescent TPE-amine 1 gradually turned off upon addition of 0–2 equiv. of nitrite ions, followed by the addition of alkaline β-naphthol after an interval of 5 min at 0–5 °C and incubation of the resultant mixture for a further 5 min (Fig. 4). Initially, the solution showed an intense blue fluorescence at λmax 447 nm and upon the addition of nitrite the fluorescence intensity decreased slowly, with almost no change in the absorption maxima. It is expected that probe 1 would be quickly diazotized in the presence of nitrite ions under acidic conditions at low temperature and would react with alkaline β-naphthol to form the corresponding azo-dye 2. Since the dye is non-fluorescent in solution as well as in the solid state, the fluorescence response decreases gradually. A drop in the fluorescence intensity of about 15-fold was observed, indicating that the probe could efficiently detect trace levels of nitrite ions in solution. The azo-dye 2 was subsequently isolated in good quantity by carrying out a relatively large scale reaction and its structure was confirmed using 1H NMR, 13C NMR and ESI-MS spectra (see ESI† for details). In a separate experiment, the non-fluorescent nature of azo-dye 2 in solution was verified by measuring its fluorescence response in various proportions of THF in water (Fig. S6 of ESI†).
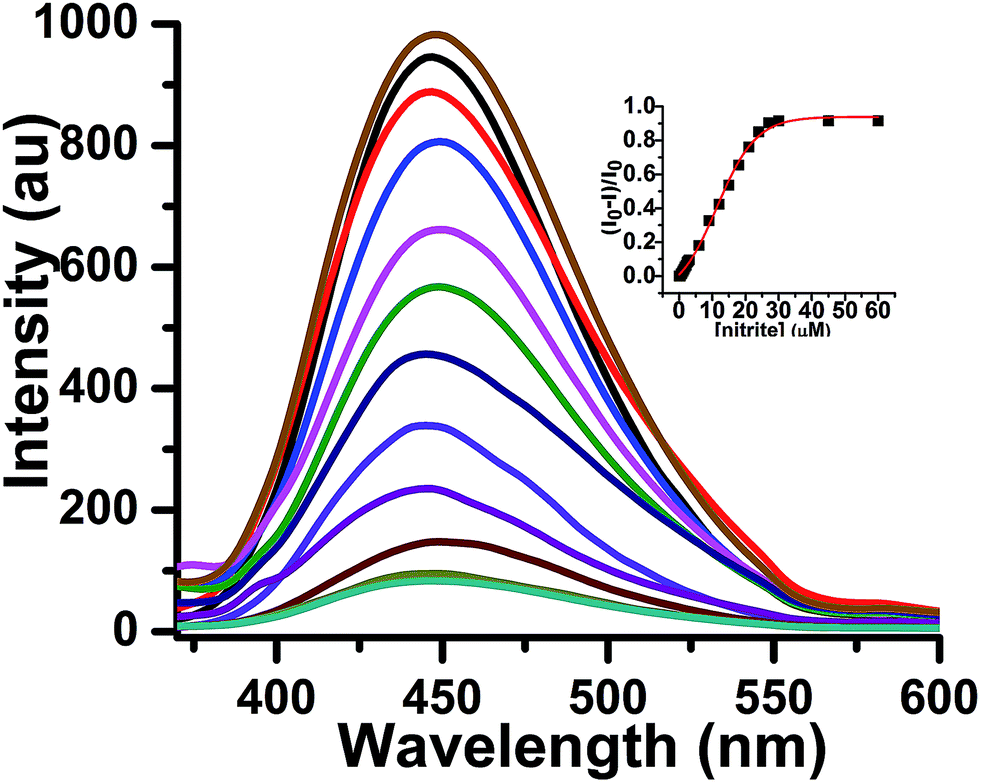 | ||
| Fig. 4 Fluorescence response of probe 1 (30 μM) upon addition of different concentrations of nitrite (0–60 μM) and alkaline β-naphthol solution [solvent system: 3% THF in 0.1 N aqueous HCl; λex 345 nm]. Inset: plot of relative emission intensity against no. of equiv. of nitrite ions. | ||
The selectivity of probe 1 was assessed by challenging it with several other environmentally relevant anions, such as I−, Br−, CH3COO−, N3−, SO4−, NO3−, which do not induce any change in fluorescence intensity relative to the blank, even at relatively high concentrations, providing evidence for its high selectivity to nitrite ions (Fig. 5).
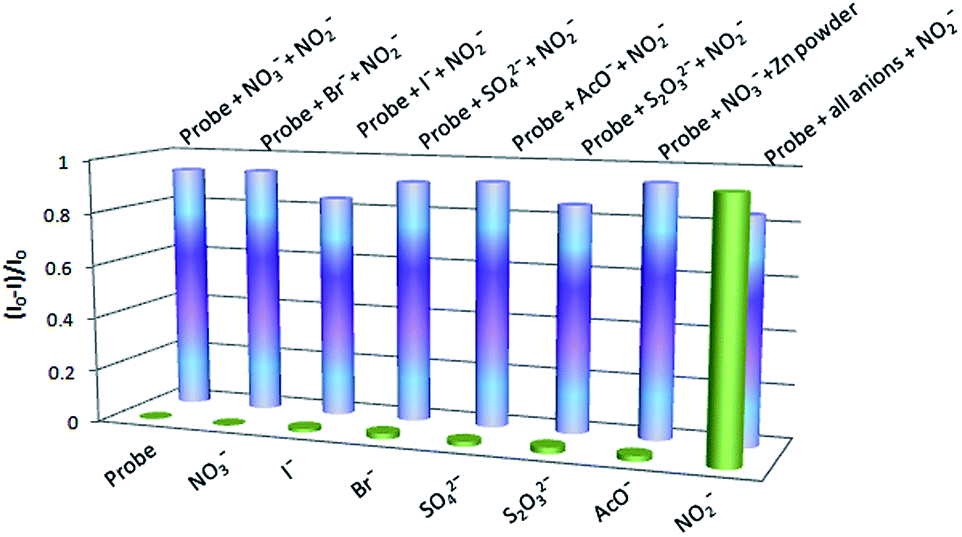 | ||
| Fig. 5 Maximum fluorescence response of probe 1 (30 μM) upon addition of different anions (30 μM) in 3% THF–0.1 N aqueous HCl. | ||
The specificity is an outcome of the fact that diazotization of the amine group of probe 1 is only possible when nitrite ions are present in the system. However, the expected drop in the fluorescence intensity was not observed in the case of oxidizable anions like I−, S2O32− and Br−. Oxidizable anions, mainly iodide, are expected to impede the detection of nitrite ions by reacting with them under acidic conditions to produce NO gas.20 However, as a reactive radical species, NO does not last long in aqueous solution but undergoes oxidation to generate mainly nitrite ions.21 Making an allowance for these facts, we presume that both the reactions take place in a successive manner, to different extents depending on the kind of oxidizable anion present in the solution for the competition experiments, maintaining a similar level of nitrite ions in solution and rendering a minor drop in the fluorescence intensity. Our conjecture was supported by the fact that the fluorescence emission of probe 1 is negligible in the presence of NO under the established diazotization conditions at pH 1 (Fig. S7 of ESI†).
Our sensing system is able to detect nitrite ions well below 1 ppm, the MCL set by the EPA for drinking water. Probe 1 responds to NO2− ions linearly below the micro molar concentration range and from that the detection limit of probe 1 was estimated to be 6 × 10−7 M or 27.6 ppb of NaNO2 (Fig. S8 of ESI†).
Notably, we extended the capability of this probe to the detection of nitrate ions by performing an in situ Zn powder-facilitated reduction step. The reduction was carried out by adding a small amount of zinc (a toothpick full), which spontaneously catalyzes the reduction of nitrate to nitrite ions in a short amount of time. Therefore, a similar fluorimetric response was obtained to that of nitrite ions when 0–2 equiv. of nitrate ions was added to probe 1 in 3% THF in 0.1 N HCl at 0–5 °C, adding a toothpick full of Zn-dust and shaking the resultant mixture for 5 min followed by addition of alkaline β-naphthol solution and incubation of the resultant mixture for a further 5 min (Fig. 6).
 | ||
| Fig. 6 Fluorescence response of probe 1 (30 μM) upon addition of different concentration of nitrate (0–60 μM) and a small amount of Zn powder, followed by alkaline β-naphthol solution [solvent system: 3% THF in 0.1 N aqueous HCl; λex 345 nm]. Inset: plot of relative emission intensity against no. of equiv. of nitrate ions. | ||
The applicability of probe 1 was verified by detecting the nitrite levels in several real samples. The samples were collected from various sources, such as river, paddy field, and aquarium water. For the determination of nitrite levels in each real sample, it was made appropriately acidic by addition of conc. HCl, then probe 1 was added at 0–5 °C followed by the addition of β-naphthol, and the fluorescence intensity was measured. The levels of nitrite in the real samples were obtained by plotting the intensity values on a standard fluorescence intensity curve and were found to be in the range of 30–60 ppb (Fig. 7).
 | ||
| Fig. 7 A plot of the relative intensities of different real samples on a standard fluorescence curve to quantify the levels of nitrite ions in those samples. | ||
Conclusions
In conclusion, we have developed a novel AIE-based fluorescent probe for the detection of trace amounts of nitrite ions in water. The function of probe 1, which is an amine derivative of tetraphenylethylene, depends on diazotization of its amino group in the presence of dissolved nitrite followed by coupling with β-naphthol to produce an azo-dye (2). Although the probe is highly fluorescent in 3% THF–0.1 N HCl solution, the resultant azo-dye is non-fluorescent in solution as well as in the solid sate and thereby, the probe acts as a turn-off type molecular sensor for nitrite ions in the presence of many other interfering anionic species. The probe works equally well for the detection of nitrate ions under reducing conditions (in the presence of Zn-dust). High sensitivity and selectivity, low detection limit, and fast reaction coupled with cost effective synthesis make this probe highly promising for practical purposes.Acknowledgements
A.C. thanks DST (India) (project no. SR/FT/CS-092/2009) for financial support. M.B. is also thankful to CSIR (India) (project no. 02(0075)/2012/EMR-II) for research funding. D.G.K. is thankful to DST (India) for research fellowships. The authors sincerely thank Dr Supriya Tilvi of NIO, Goa, India for timely help with some spectral data.Notes and references
- C. J. Johnson and C. K. Burton, Am. J. Ind. Med., 1990, 18, 449 CrossRef CAS.
- M. T. Gladwin, A. N. Schechter, D. B. Kim-Shapiro, R. P. Patel, N. Hogg, S. Shiva, R. O. Cannon III, M. Kelm, D. A. Wink, M. G. Espey, E. H. Oldfield, R. M. Pluta, B. A. Freeman, J. R. Lancaster Jr, M. Feelisch and J. O. Lundberg, Nat. Chem. Biol., 2005, 1, 308 CrossRef CAS PubMed.
- I. A. Wolff and A. E. Wasserman, Science, 1972, 177, 15 CAS.
- J. O. Lundberg, E. Weitzberg and M. T. Gladwin, Nat. Rev. Drug Discovery, 2008, 7, 156 CrossRef CAS PubMed.
- (a) G. J. Mohr and O. S. Wolfbeis, Analyst, 1996, 121, 1489 RSC; (b) M. Bru, M. I. Burguete, F. Galindo, S. V. Luis, M. J. Marín and L. Vigara, Tetrahedron Lett., 2006, 47, 1787 CrossRef CAS PubMed.
- (a) P. Wang, X. Wang, L. Bi and G. Zhu, Analyst, 2000, 125, 1291 RSC; (b) A. Rahim, L. S. S. Santos, S. B. A. Barros, L. T. Kubota, R. Landers and Y. Gushikem, Electroanalysis, 2014, 26, 541 CrossRef CAS.
- J. M. Doyle, M. L. Miller, B. R. McCord, D. A. McCollam and G. W. Mushrush, Anal. Chem., 2000, 72, 2302 CrossRef CAS.
- (a) X. Chen, F. Wang and Z. Chen, Anal. Chim. Acta, 2008, 623, 213 CrossRef CAS PubMed; (b) Y. Qiu, H. Deng, J. Mou, S. Yang, M. Zeller, S. R. Batten, H. Wu and J. Li, Chem. Commun., 2009, 5415 RSC; (c) P. Li, Y. Ding, A. Wang, L. Zhou, S. Wei, Y. Zhou, Y. Tang, Y. Chen, C. Cai and T. Lu, ACS Appl. Mater. Interfaces, 2013, 5, 2255 CrossRef CAS PubMed.
- (a) W. L. Daniel, M. S. Han, J.-S. Lee and C. A. Mirkin, J. Am. Chem. Soc., 2009, 131, 6362 CrossRef CAS PubMed; (b) N. Adarsh, M. Shanmugasundaram and D. Ramaiah, Anal. Chem., 2013, 85, 10008 CrossRef CAS PubMed; (c) N. Xiao and C. Yu, Anal. Chem., 2010, 82, 3659 CrossRef CAS PubMed.
- (a) Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192 CrossRef CAS PubMed; (b) R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936 RSC; (c) M. J. Culzoni, A. M. de la Pena, A. Machuca, H. C. Goicoechea and R. Babiano, Anal. Methods, 2013, 5, 30 RSC and references cited therein (d) H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210 RSC; (e) M. E. Jun, B. Roy and K. H. Ahn, Chem. Commun., 2011, 47, 7583 RSC; (f) F. Long, A. Zhu, H. Shi, H. Wang and J. Liu, Sci. Rep., 2013, 3, 2308 Search PubMed; (g) M. Li, H. Gou, I. Al-Ogaidi and N. Wu, ACS Sustainable Chem. Eng., 2013, 1, 713 CAS; (h) J. Chan, S. C. Dodanil and C. J. Chang, Nat. Chem., 2012, 4, 973 CrossRef CAS PubMed; (i) X. Chen, G. Zhou, X. Peng and J. Yoon, Chem. Soc. Rev., 2012, 41, 4610 RSC.
- (a) V. Kumar, M. Banerjee and A. Chatterjee, Talanta, 2012, 99, 610 CrossRef CAS PubMed; (b) A. Büldt and U. Karst, Anal. Chem., 1999, 71, 3003 CrossRef; (c) M. Strianese, S. Milione, V. Bertolasi and C. Pellecchia, Inorg. Chem., 2013, 52, 11778 CrossRef CAS PubMed.
- (a) J. B. Brirks, Photophysics of Aromatic Molecules, Wiley, London, 1970 Search PubMed; (b) H. Tong, Y. Hong, Y. Dong, M. Haubler, J. W. Y. Lam, Z. Li, Z. Guo, Z. Guo and B. Z. Tang, Chem. Commun., 2006, 3705 RSC.
- B. Z. Tang and A. Qin, Aggregation-Induced Emission: Fundamentals and Applications, Wiley, New York, 2013, vol. 1 and 2 Search PubMed.
- For selected recent reviews on AIE-active materials and their applications, see: (a) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed; (b) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878 RSC; (c) Y. Hong, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (d) H. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC; (e) R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015 10.1039/c4cs00325j.
- For some recent examples on TPE-based chemosensors/bioprobes, see: (a) H.-T. Feng, S. Song, Y.-C. Chen, C.-H. Shen and Y.-S. Zheng, J. Mater. Chem. C, 2014, 2, 2353 RSC; (b) J. Zhang, Q. Yang, Y. Zhu, H. Liu, Z. Chi and C.-Y. Su, Dalton Trans., 2014, 43, 15785 RSC; (c) N. Zhao, J. W. Y. Lam, H. H. Y. Sung, H. M. Su, I. D. Williams, K. S. Wong and B. Z. Tang, Chem.–Eur. J., 2014, 20, 133 CrossRef CAS PubMed; (d) X. Wang, J. Hu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2014, 136, 9890 CrossRef CAS PubMed; (e) H.-T. Feng and Y.-S. Zheng, Chem.–Eur. J., 2014, 20, 195 CrossRef CAS PubMed; (f) X. Lou, C. W. T. Leung, C. Dong, Y. Hong, S. Chen, E. Zhao, J. W. Y. Lam and B. Z. Tang, RSC Adv., 2014, 4, 33307 RSC; (g) X. Wang, H. Liu, J. Li, K. Ding, Z. Lv, Y. Yang, H. Chen and X. Li, Chem.–Asian J., 2014, 9, 784 CrossRef CAS PubMed; (h) C. J. Kassl and F. C. Pigge, Tetrahedron Lett., 2014, 55, 4810 CrossRef CAS PubMed; (i) J.-H. Ye, J. Liu, Z. Wang, Y. Bai, W. Zhang and W. He, Tetrahedron Lett., 2014, 55, 3688 CrossRef CAS PubMed; (j) F. Hu, Y. Huang, G. Zhang, R. Zhao and D. Zhang, Tetrahedron Lett., 2014, 55, 1471 CrossRef CAS PubMed; (k) T. Noguchi, B. Roy, D. Yoshihara, Y. Tsuchiya, T. Yamamoto and S. Shinkai, Chem.–Eur. J., 2014, 20, 381 CrossRef CAS PubMed; (l) D. G. Khandare, V. Kumar, A. Chattopadhyay, M. Banerjee and A. Chatterjee, RSC Adv., 2013, 3, 16981 RSC; (m) D. G. Khandare, H. Joshi, M. Banerjee, M. S. Majik and A. Chatterjee, RSC Adv., 2014, 4, 47076 RSC; (n) J. Mei, Y. Wang, J. Tong, J. Wang, A. Qin, J. Z. Sun and B. Z. Tang, Chem.–Eur. J., 2013, 19, 613 CrossRef CAS PubMed; (o) J. Li, J. Liu, J. W. Y. Lam and B. Z. Tang, RSC Adv., 2013, 3, 8193 RSC; (p) J. Liang, R. T. K. Kwok, H. Shi, B. Z. Tang and B. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 8784 CrossRef CAS PubMed; (q) C. Yu, Y. Wu, F. Zeng, X. Li, J. Shi and S. Wu, Biomacromolecules, 2013, 14, 4507 CrossRef CAS PubMed; (r) H. Liu, Z. Lv, K. Ding, X. Liu, L. Yuan, H. Chen and X. Li, J. Mater. Chem. B, 2013, 1, 5550 RSC; (s) Y. Yan, Z. Che, X. Yu, X. Zhi, J. Wang and H. Xu, Bioorg. Med. Chem., 2013, 21, 508 CrossRef CAS PubMed; (t) T. Han, X. Feng, B. Tong, J. Shi, L. Chen, J. Zhi and Y. Dong, Chem. Commun., 2012, 48, 416 RSC; (u) G. Huang, G. Zhang and D. Zhang, Chem. Commun., 2012, 48, 7504 RSC; (v) X. Wang, J. Hu, T. Liu, G. Zhang and S. Liu, J. Mater. Chem., 2012, 22, 8622 RSC; (w) Y. Liu, Z. Wang, G. Zhang, W. Zhang, D. Zhang and X. Jiang, Analyst, 2012, 137, 4654 RSC.
- For some recent examples of TPE-based molecules for various other applications, see: (a) Y. Chen, M. Li, Y. Hong, J. W. Y. Lam, Q. Zheng and B. Z. Tang, ACS Appl. Mater. Interfaces, 2014, 6, 10783 CrossRef CAS PubMed; (b) C. Zhang, S. Jin, S. Li, X. Xue, J. Liu, Y. Huang, Y. Jiang, W.-Q. Chen, G. Zou and X.-J. Liang, ACS Appl. Mater. Interfaces, 2014, 6, 5212 CrossRef CAS PubMed; (c) S. Yao, X. Yang, M. Yu, Y. Zhang and J.-X. Jiang, J. Mater. Chem. A, 2014, 2, 8054 RSC; (d) W. Qin, K. Li, G. Feng, M. Li, Z. Yang, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2014, 24, 635 CrossRef CAS; (e) C. W. T. Leung, Y. Hong, S. Chen, E. Zhao, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 62 CrossRef CAS PubMed; (f) Z. Zhao, C. Y. K. Chan, S. Chen, C. Deng, J. W. Y. Lam, C. K. W. Jim, Y. Hong, P. Lu, Z. Chang, X. Chen, P. Lu, H. S. Kwok, H. Qiu and B. Z. Tang, J. Mater. Chem., 2012, 22, 4527 RSC; (g) J.-P. Xu, Y. Fang, Z.-G. Song, J. Mei, L. Jia, A. Qin, J. Z. Sun, J. Ji and B. Z. Tang, Analyst, 2011, 136, 2315 RSC.
- For selected recent examples of various AIE-active materials other than TPE and their applications, see: (a) N. Na, F. Wang, J. Huang, C. Niu, C. Yang, Z. Shang, F. Han and J. Ouyang, RSC Adv., 2014, 4, 35459 RSC; (b) L. Wang, D. Wang, H. Lu, H. Wang, L. Xue and S. Feng, Appl. Organomet. Chem., 2013, 27, 529 CrossRef CAS; (c) O. Simalou, R. Lu, P. Xue, P. Gong and T. Zhang, Eur. J. Org. Chem., 2014, 2014, 2907 CrossRef CAS; (d) G. Zhang, A. Ding, Y. Zhang, L. Yang, L. Kong, X. Zhang, X. Tao, Y. Tian and J. Yang, Sens. Actuators, B, 2014, 202, 209 CrossRef CAS PubMed; (e) C. Y. K. Chan, J. W. Y. Lam, Z. Zhao, S. Chen, P. Lu, H. H. Y. Sung, H. S. Kwok, Y. Ma, I. D. Williams and B. Z. Tang, J. Mater. Chem. C, 2014, 2, 4320 RSC; (f) X. Tang, L. Yao, H. Liu, F. Shen, S. Zhang, H. Zhang, P. Lu and Y. Ma, Chem.–Eur. J., 2014, 20, 7589 CrossRef CAS PubMed; (g) H. Zhang, Y. Qu, Y. Gao, J. Hua, J. Li and B. Li, Tetrahedron Lett., 2013, 54, 909 CrossRef CAS PubMed; (h) W. Z. Yuan, Y. Gong, S. Chen, X. Y. Shen, J. W. Y. Lam, P. Lu, Y. Lu, Z. Wang, R. Hu, N. Xie, H. S. Kwok, Y. Zhang, J. Z. Sun and B. Z. Tang, Chem. Mater., 2012, 24, 1518 CrossRef CAS; (i) X. Zhang, X. Zhang, S. Wang, M. Liu, L. Tao and Y. Wei, Nanoscale, 2013, 5, 147 RSC; (j) J. Mei, J. Wang, J. Z. Sun, H. Zhao, W. Yuan, C. Deng, S. Chen, H. H. Y. Sung, P. Lu, A. Qin, H. S. Kwok, Y. Ma, I. D. Williams and B. Z. Tang, Chem. Sci., 2012, 3, 549 RSC; (k) Y. Liu, Y. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Y. Lam, R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2010, 132, 13951 CrossRef CAS PubMed; (l) L. Xu, Y. Li, S. Li, R. Hu, A. Qin, B. Z. Tang and B. Su, Analyst, 2014, 139, 2332 RSC.
- S. Hazra, S. Balaji, M. Banerjee, A. Ganguly, N. N. Ghosh and A. Chatterjee, Anal. Methods, 2014, 6, 3784 RSC.
- X. F. Duan, J. Zeng, J. W. Lu and Z. B Zhang, J. Org. Chem., 2006, 71, 9873 CrossRef CAS PubMed.
- C. A. Abeledo and I. M. Kolthoff, J. Am. Chem. Soc., 1931, 53, 2893 CrossRef CAS.
- L. J. Ignarro, J. M. Fukuto, J. M. Griscavage, N. E. Rogers and R. E. Byrns, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 8103 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General information, synthetic procedures, experimental procedures, pH study, limit of detection, details of theoretical calculations and spectral data; spectra of probe 1 and azo dye 2. See DOI: 10.1039/c4ra14765k |
| This journal is © The Royal Society of Chemistry 2015 |