
A sustainable iron-based sodium ion battery of porous carbon–Fe3O4/Na2FeP2O7 with high performance
Jun Ming†
*a,
Hai Ming†b,
Wenjing Yangc,
Won-Jin Kwaka,
Jin-Bum Parka,
Junwei Zhengb and
Yang-Kook Sun*a
aDepartment of Energy Engineering, Hanyang University, Seoul 133-791, South Korea. E-mail: yksun@hanyang.ac.kr; mingjun6297@gmail.com; Fax: +82 2 2282 7329; Tel: +82 2 2220 0524
bCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, P. R. China
cReliability Research and Analysis Center, CEPREI (East China) Laboratories, The Fifth Research Institute of MIIT East China, P. R. China
First published on 15th December 2014
Abstract
A type of porous carbon–Fe3O4 (e.g., PC–Fe3O4) composite with an industrially scalable production was introduced in the sodium ion battery application for the first time. The PC–Fe3O4 composite, consisting of highly dispersed Fe3O4 nanocrystals within the porous carbon with a relatively low weight percent of 45.5 wt%, could efficiently demonstrate high capacities of 225, 168, 127, 103, 98 and 90 mA h g−1 under the current densities of 50, 100, 200, 300, 400 and 500 mA g−1 with a good stability over 400 cycles. The utilization co-efficient of Fe3O4 nanocrystals was proven to be much higher than most of the Fe3O4 nanoparticles reported recently via the study of the capacity contribution of carbon originally. In addition, the robustness of electrode during the charge–discharge was well characterized by ex situ XRD and emission scanning electron microscopy (SEM). More importantly, a new concept of an elemental iron-based sodium ion battery of PC–Fe3O4/Na2FeP2O7 is presented. This is the first example to introduce an element-rich configuration in the sodium ion battery from the viewpoint of sustainability. The full battery demonstrated a superior capacity of 93 mA h g−1, high capacity retention of 93.3% over 100 cycles and work voltage around 2.28 V with the energy density of 203 W h kg−1. Such configuration of an iron-based sodium battery would be highly promising and sustainable owing to its low cost and high stability in grid storage.
Introduction
The development of cost-effective batteries with high safety is a great challenge for promising large-scale application of lithium and sodium batteries in electronic vehicle (EV) and grid storage.1–3 In particular, with the strong tendency of abundant sodium ion batteries, instead of the source/cost-limited lithium one, the safety issue becomes ever more important due to the higher burning activity of sodium metal exposed to oxygen or moisture.4 Considering these aspects, the configuration of ideal sodium ion battery system, particularly based on developing an appropriate anode and cathode, is very significant to ensure its sustainability and safety.To develop the anode, a popular research trend of preparing various carbon and/or metal (oxide) based materials appeared recently in the similar way as those in the lithium ion battery.5–9 Carbon and metal oxide-based materials with different morphologies, structures and compositions are being widely synthesized such as carbon fibers,10 hollow carbon tubes,11 graphene,12 Sn–SnS–C,13 Fe2O3–graphene,14 CuO arrays,15 TiO2 (ref. 16) and TiO2–C.17 Indeed, most of them demonstrated excellent performance due to the intriguing properties of nano-characteristics. However, the aspects of cost and safety of materials, as well as the synthetic process should be well considered for their practical commercialization. To improve the safety, more and more researchers prefer to develop metal oxide-based anodes instead of carbon because of their great advantages of high capacity, non-flammable ability and low voltage of around 0.7–0.9 V, which could effectively suppress the deposition of metal dendrites.18 However, inevitably, most processes always spend a high cost and need complex procedures for the manipulation. Naturally, it would be great to obtain nano-structured materials with a high performance via a simple approach, particularly with using low-cost elements that are abundant in the earth. Therefore, we introduce herein a new composite iron oxide-based anode of porous carbon–Fe3O4 in the sodium ion battery for the first time, which could be obtained in an industrially scalable way, rather than the experimental amount of Fe3O4 particles obtained by the hydrothermal method.19–21 It is well known that iron is the fourth highest element in the earth and is only less than O, Si and Al. The high content of 5.1 wt%, together with the physicochemical characteristics of highly non-flammable and a suitable voltage around 0.7–0.9 V, could make the iron oxide-based anode to be a very competitive anode in batteries for sustainability.
Although the sodium ion battery has been attracting great attention recently both for anodes and cathodes (e.g., P2-type Nax[Fe1/2Mn1/2]O2,22 Nax[Ni1/3Mn2/3]O2,23 Na3V2(PO4)2F3,24 Na2FeP2O7 (ref. 25 and 26)), the cases of developing a full battery are very limited,27 especially further concerning the matters of safety and cost. Undoubtedly, it would be very interesting and significant to develop full batteries to evaluate their combined performance as those in practical applications, and it should be a big step towards commercialization, which is an improvement from the isolated research on anode or cathode. To accelerate the availability of these materials and their utilization in practical applications, herein we introduce a new concept of elemental iron-based sodium battery PC–Fe3O4/Na2FeP2O7, in which the safe and cost-effective electrode Na2FeP2O7 and PC–Fe3O4 were chosen. It is the first example to introduce an element-rich configuration in a sodium ion battery from the viewpoint of sustainability. The designed battery demonstrated a high capacity of 93 mA h g−1 at 0.1 C, cycle ability over 100 cycle with a capacity retention of 93.3% at 0.1 C, work voltage around 2.28 V with an energy density of 203 W h kg−1. In theory, such configuration of an elemental iron-based battery is highly promising and sustainable because of its low cost and high safety.
Experimental
Anode preparation
First, 15.0 g of Pluronic F127 was dissolved in 60 g of ethanol at 40 °C, and then 25 g of Fe(NO3)3·9H2O and 45 g of resol–ethanol (20 wt %) were added into the solution successively. After stirring for 4 h, the solution was then transferred into an oven and dried at a high temperature of 100 °C. The heating rate was controlled to be slow till the final 100 °C to avoid any mild explosions of solution. The dried sample was calcined at 500 °C (heating rate, 2 °C min−1) for 2 h in the furnace under an Ar flow. Without the addition of precursor Fe(NO3)3·9H2O into the solution, porous carbon could be obtained under the same procedure. For comparison, Fe3O4 particles were prepared according to previously described procedures.28,29 Typically, 27 g of FeCl3·6H2O was dissolved in 800 ml ethylene glycol to form a clear solution, and then 72 g of NaAc and 20 g of polyethylene glycol (PEG-400) were added in the solution. The mixture was vigorously stirred for 30 min and then sealed in a 1000 ml Teflon lined stainless-steel autoclave. The autoclave was heated to and maintained at 200 °C for 8 h, and then allowed to naturally cool to room temperature. The Fe3O4 particles were washed with water and ethanol several times with stirring. It should be noted that the samples could be effectively collected by a magnet after each washing; thus, any separation technique or centrifugation was not required.Cathode preparation
Na2FeP2O7 material was synthesized via a solid state reaction, in which the stoichiometric amounts of Na2CO3, (NH4)2HPO4 and FeC2O4·2H2O were ball milled first.30 Then, the powder was pelletized and pre-heated at 350 °C for 3 h in an argon atmosphere. The pellet was reground into powder, and then re-pelletized for re-calcination at 600 °C for 6 h in Ar. Finally, the obtained Na2FeP2O7 was further coated by the carbon based on the following procedure. The powders of Na2FeP2O7 and acetylene black (AB) with the mass ratios of 8/2 were ball-milled, and then the calcination of the pressed pellet at 600 °C for 10 h under Ar flow produced carbon modified Na2FeP2O7.Electrode preparation
The anode PC–Fe3O4, the binder CMC/PAA rather than polyvinylidene difluoride (PVDF), and the conductive carbon AB with a mass ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 were mixed in water to form a homogeneous slurry. Then, the slurry was cast on a copper foil by a doctor blade. It was kept at room temperature till it was almost dry and then transferred to a vacuum oven and dried at 80 °C overnight. The foil was punched to form a circular electrode with the diameter of 16 mm. The mass density of PC–Fe3O4 in the electrode was about 1.5 mg cm−2. Alternatively, the powder of the electrode, obtained from vacuum-drying the aqueous slurry of PC–Fe3O4–CMC/PAA–AB, was pressed into a tablet (Ø 14 mm) for the characterization by ex situ XRD with the aim of avoiding the interference of copper foil. The reason for selecting the binder CMC/PAA and the green solvent water were ascribed to the better performance in lithium and sodium ion battery applications as reported recently.31 For the preparation of the cathode, composite Na2FeP2O7–AB, binder PVDF, and conductive carbon AB, were mixed well in the mass ratio of 85:10:5 to form a slurry, and then it was casted on an Al foil. It was then dried at 120 °C in a vacuum oven overnight and then punched into a circular electrode before use. The mass density of Na2FeP2O7 in the electrode was around 3.48 mg cm−2.
:10:10 were mixed in water to form a homogeneous slurry. Then, the slurry was cast on a copper foil by a doctor blade. It was kept at room temperature till it was almost dry and then transferred to a vacuum oven and dried at 80 °C overnight. The foil was punched to form a circular electrode with the diameter of 16 mm. The mass density of PC–Fe3O4 in the electrode was about 1.5 mg cm−2. Alternatively, the powder of the electrode, obtained from vacuum-drying the aqueous slurry of PC–Fe3O4–CMC/PAA–AB, was pressed into a tablet (Ø 14 mm) for the characterization by ex situ XRD with the aim of avoiding the interference of copper foil. The reason for selecting the binder CMC/PAA and the green solvent water were ascribed to the better performance in lithium and sodium ion battery applications as reported recently.31 For the preparation of the cathode, composite Na2FeP2O7–AB, binder PVDF, and conductive carbon AB, were mixed well in the mass ratio of 85:10:5 to form a slurry, and then it was casted on an Al foil. It was then dried at 120 °C in a vacuum oven overnight and then punched into a circular electrode before use. The mass density of Na2FeP2O7 in the electrode was around 3.48 mg cm−2.
Characterization
The morphology and structure of PC–Fe3O4 and electrodes were characterized by field emission scanning electron microscopy (FESEM), which were obtained on an XL30 ESEM microscope with a beam energy of 20 kV. The encapsulated Fe3O4 nanocrystals were characterized by transmission electron microscopy (TEM) images using a JEOL-2100F microscope operated at 200 kV. The crystallographic information of PC–Fe3O4 and the electrodes were investigated by XRD, which was measured on a Brucker D8 GADDS diffractometer using Co Kα radiation (1.79 Å). For the cycled electrodes disassembled from the coin cell, they were protected by a cover to avoid contact with air. The valencies of Fe were determined using XPS, and the spectrum was recorded on an ESCALAB 250 spectrometer. The monochromatized Al Kα X-ray source for the XPS was operated at 12 kV and 20 mA.32 The surface area and porosity of PC–Fe3O4 and porous carbon were determined using a Quantachrome Autosorb-1-MP automated gas adsorption system using nitrogen as the adsorbate at liquid nitrogen temperature (77 K). The samples were out-gassed under vacuum for 24 h at 100 °C. The specific surface area was calculated using the Brunauer–Emmett–Teller (BET) method, and the porous characteristic was determined using the t-plot method. The mass content of Fe3O4 was measured by thermal gravimetric analysis (TGA, 10 °C min−1, Air). The 2032-type coin cells were assembled in a glove box in which both the water and oxygen content were below 0.1 ppm. In the battery test, the electrolyte was 1.0 M NaClO4 in propylene carbonate (PC) with 2 vol% fluoroethylene carbonate (FEC), and a glass fibre was used as the separator. The PC–Fe3O4/Na and PC–Fe3O4/Na2FeP2O7 batteries were tested within the voltage range of 0.01–3 V and 1.1–4.2 V, respectively using a TOCAST 3100 instrument at a temperature of 30 °C. The cyclic voltammetry (CV) of Fe3O4/Na within the voltage range of 0.01–3.0 V was analyzed using a VMP-3 instrument.Results and discussion
The hierarchical structure of PC–Fe3O4 composite, consisting of encapsulated Fe3O4 nanocrystals with the size of around 10–15 nm within the porous carbon, can be clearly observed and confirmed by the SEM, TEM and HRTEM images (Fig. 1a–c, marked by yellow circle). The exposed lattice distance of 2.518 Å correspond well to the Fe3O4 planes of (311) (Fig. 1c). In addition, the indexed diffraction patterns of (002), (311), (004), (333) and (044) in the selected area electron diffraction (SAED) were in good accordance with the XRD result, as shown in Fig. 2a, except for the pattern of (422), which was too weak to be observed in the SAED and also not obvious in the XRD (Fig. 1d and 2a). The crystal structure of Fe3O4 belongs to the space group of F![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) 3mz (ICSD#633020),33 in which the octahedral FeO6 were corner connected by bridged O and the cell parameters of a, b and c were 8.3528 Å, as characterized by the Rietveld results of the XRD pattern (Fig. 2b).34 The main visual structure of PC–Fe3O4 is shown in Fig. 1d, in which the Fe3O4 nanocrystals are embedded and highly dispersed in the porous carbon. The valency of iron was further confirmed by XPS. As shown in Fig. 3a, the spin–orbit split Fe2p peaks are broad due to a small chemical shift between Fe2+ and Fe3+, both of which are present in Fe3O4. The Fe2+ and Fe3+ components were determined by fitting the spectral line shapes to a convolution of Gaussian and Lorentzian functions. The obtained Fe2p2/3 (2p1/2) binding energy is 710.6 eV (725.0 eV) for Fe2+ and 711.9 eV (723.6 eV) for Fe3+. These values efficiently match the literature values.35–37 The mass percent of Fe3O4 was about 44.5% and the surface area of the composite could be as high as 200 m2 g−1 with a porosity of around 5.9 nm and rich volume of 0.26 cm3 g−1 (Fig. 3b and c). The rich porosity undoubtedly would facilitate the permeation of electrolyte into the pores for a good transfer ability of lithium or sodium ions.38
3mz (ICSD#633020),33 in which the octahedral FeO6 were corner connected by bridged O and the cell parameters of a, b and c were 8.3528 Å, as characterized by the Rietveld results of the XRD pattern (Fig. 2b).34 The main visual structure of PC–Fe3O4 is shown in Fig. 1d, in which the Fe3O4 nanocrystals are embedded and highly dispersed in the porous carbon. The valency of iron was further confirmed by XPS. As shown in Fig. 3a, the spin–orbit split Fe2p peaks are broad due to a small chemical shift between Fe2+ and Fe3+, both of which are present in Fe3O4. The Fe2+ and Fe3+ components were determined by fitting the spectral line shapes to a convolution of Gaussian and Lorentzian functions. The obtained Fe2p2/3 (2p1/2) binding energy is 710.6 eV (725.0 eV) for Fe2+ and 711.9 eV (723.6 eV) for Fe3+. These values efficiently match the literature values.35–37 The mass percent of Fe3O4 was about 44.5% and the surface area of the composite could be as high as 200 m2 g−1 with a porosity of around 5.9 nm and rich volume of 0.26 cm3 g−1 (Fig. 3b and c). The rich porosity undoubtedly would facilitate the permeation of electrolyte into the pores for a good transfer ability of lithium or sodium ions.38
 | ||
| Fig. 1 (a) SEM, (b) TEM, (c) HRTEM and (d) SAED of PC–Fe3O4 composite. | ||
 | ||
| Fig. 2 (a) Rietveld refinement of the XRD pattern of PC–Fe3O4 (Rwp = 1.073%, GOF = 1.127). The theoretical Bragg positions are shown with green ticks. (b) Crystalline structure of Fe3O4 along c-axis. (c) Schematic structure of PC–Fe3O4 composite. Inset: green crystallites are Fe3O4 nanocrystals, black areas are carbon, blank channels are pore channels. | ||
 | ||
| Fig. 3 (a) XPS of Fe2p, (b) TGA, and BET analysis of (c) PC–Fe3O4 and (d) porous carbon. Insets of (c) and (d) are pore size distribution of PC–Fe3O4 and porous carbon respectively. | ||
The reason for introducing the Fe3O4 oxide into the carbon matrix should be ascribed to the high conductive ability and protection of carbon for Fe3O4 in the repeated charge and discharge cycles; this was also confirmed from many studies previously reported in the literature (e.g., Fe3O4@C (ref. 39) and Fe3O4@CFx (ref. 40)). However, the contribution of carbon to the capacity of the composite was not studied individually before. Herein, we, for the first time, investigated its effect in the sodium ion battery, and an interesting phenomena could be observed. As shown in Fig. 4a, a large irreversible capacity exists in the PC–Fe3O4 composite, in which a high capacity of 672 mA h g−1 was obtained in the 1st cycle but only 252 mA h g−1 was delivered in the 2nd cycle. To explore the origin of the large irreversible capacity, the electrochemical performance of porous carbon and neat Fe3O4 were investigated.
 | ||
| Fig. 4 Typical (a and b) rate and (c and d) cycling performance of PC–Fe3O4 in a sodium ion battery versus sodium metal. Inset of (a) and (b) are CV and dQ–V/V plots, respectively. | ||
The porous carbon, obtained from the carbonization of resin under the same process, has a very high surface area of 552 m2 g−1, uniform porosity of 1.9 nm, and rich volume of 0.56 cm3 g−1 (Fig. 3d and 5a). In the sodium ion battery, it showed a high capacity of 500 mA h g−1 in the 1st cycle but delivered a very limited capacity of 10 mA h g−1 from the 2nd cycle and even decayed very fast in the following cycles close to 0 mA h g−1 (Fig. 5c). In contrast, the irreversibility of the Fe3O4 was considerably smaller (Fig. 5b). Without any carbon modification, the neat Fe3O4 particles delivered a capacity of 230 mA h g−1 in the 1st cycle, which decreased to 130 mA h g−1 in the 2nd cycle; it then decayed gradually to 93 mA h g−1 in the initial 50 cycles (Fig. 5d). According to the main peak in the CV around 0.9 V (inset of Fig. 4a and b), the large irreversibility should be ascribed to the formation of a solid electrolyte interface (SEI) on the carbon surface in the first cycle.41–43 It is clear that the carbon could increase the electronic conductivity and protect the structure of the metal oxide, but it has a very limited capacity contribution in this type of composite and causes a large irreversibility.
 | ||
| Fig. 5 SEM and inset TEM images of (a) porous carbon and Fe3O4 particles. (c) Voltage vs. capacity profile of (c) porous carbon and (d) Fe3O4 particles obtained by the hydrothermal method. | ||
However, compared to the neat Fe3O4 particles, PC–Fe3O4 composite demonstrated a higher and stable sodium storage ability. Under the rate test, the average capacities of 225, 168, 127, 103, 98 and 90 mA h g−1 could be obtained under the rates of 50, 100, 200, 300, 400 and 500 mA g−1. After the high rate test, the average capacity of the cell could be well recovered to 218 mA h g−1 at the current density of 50 mA g−1 (Fig. 4b). However, the cell could still work well with further cycling under the rates of 50, 100, 200, 300 mA g−1 for more than 400 cycles (Fig. 4c and d). These results demonstrate the good stability of PC–Fe3O4 as an electrode, which was mainly ascribed to the protection ability of the porous carbon from pulverization. Note that the capacity of PC–Fe3O4 was also higher than that of recently reported Fe3O4–C (<200 mA h g−1).19 Although Fe3O4 particles obtained by the hydrothermal method have been investigated as an anode in the sodium ion battery recently,19–21 the distinctive advantages of this type of PC–Fe3O4 are the industrial production and equal or even better performance, particularly with a relative low loading of Fe3O4 under which the utilization co-efficient of Fe3O4 could be largely improved.
To further evaluate the effect of carbon, a composite of Fe3O4–C composite with 65 wt% of Fe3O4 was also prepared using half the amount of resin. However, its average capacity of 104 mA h g−1 at 100 mA g−1 in the initial 50 cycles was considerably lower than 168 mA h g−1 of PC–Fe3O4 composite (Fig. 4c and 6). This should be ascribed to the aggregated Fe3O4 within the carbon, which could directly reduce the utilization co-efficient of Fe3O4. However, the high capacity, coulombic efficiency of 97.5%, and high capacity retention of 90% over 50 cycles (vs. the capacity of the 3rd cycles) of Fe3O4–C were still considerably better than the neat Fe3O4 (e.g., capacity retention of 71.5%) when 35% carbon was introduced in the composite. Note that the reduced capacity of 384 mA h g−1 (vs. 672 mA h g−1 of PC–Fe3O4 in Fig. 4a) in the first cycle further confirmed that the irreversible capacity mainly resulted from the porous carbon due to the low percent of carbon in the composite. In brief, the introduction of carbon could maintain the cycle ability of Fe3O4, but an appropriate amount of carbon is necessary to maintain its utilization co-efficient.
 | ||
| Fig. 6 (a) Discharge–charge curves and (b) cycling performances of Fe3O4–C composites at 100 mA g−1. Inset of (a) is the TGA of Fe3O4–C composite with a mass percent of 65 wt% Fe3O4. | ||
The variation of Fe3O4 in the discharge–charge process was characterized by ex situ XRD (Fig. 7). The intensity of the (004) peak increased after the milling and fabrication of the electrode due to the variation of the exposed crystal planes, but it still showed the pattern for Fe3O4. It could be found that the peaks change regularly during the discharge and charge based on the following reaction: Fe3O4 + 8Na+ + 8e− ↔ 3Fe + 4Na2O.20 Judging from Fig. 4, the actual capacity of 225 mA h gcomposite−1 (e.g., 506 mA h gFe3O4−1 versus 44.5 wt% of Fe3O4 in the PC–Fe3O4 composite) was lower than the theoretical value of 926 mA h gFe3O4−1,20 demonstrating that almost half of the Fe3O4 could react with the sodium metal. The electrode after each discharge still show the peaks of Fe3O4, which was in good accordance with the capacity as discussed. The increased density of Fe3O4 peaks after each charge could be ascribed to the following reaction: 3Fe + 4Na2O → Fe3O4 + 8Na+ + 8e−, but the peaks of Fe were not obvious (Fig. 7). This phenomenon is very similar to the one we recently reported in a lithium ion battery.44 However, the reversible variations of the electrode confirmed the stability of the electrode in the discharge and charge. In addition, compared to 130 mA h g−1 of neat Fe3O4, the utilization coefficient of Fe3O4 in PC–Fe3O4 was 54.6% (e.g., 506 mA h g−1/926 mA h g−1), which is much higher than 14.0% (e.g., 130 mA h g−1/926 mA h g−1) of Fe3O4 particles. A high capacity could be obtained with a low amount of Fe3O4, and this confirmed that the dispersion of Fe3O4 nanocrystals into the porous carbon could largely increase the utilization of metal oxide.
 | ||
| Fig. 7 Ex situ XRD of pristine and cycled electrode. | ||
Although it is a conversion mechanism with a large volume variation, the stability of the electrode efficiently demonstrated the positive effect of carbon to protect the structure in the cycling. Except for the XRD, the robustness of the electrode was further characterized by SEM. As shown in Fig. 8, the bulky particles of PC–Fe3O4 in the electrode were completely preserved after discharge and charge (Fig. 8a, c and e). After the 1st discharge, we found that a thick solid electrolyte interface layer was covered on the pristine PC–Fe3O4 particles, making the porous surface smooth (Fig. 8a–d). The observed SEI resulted from the decomposition of the electrolyte and side reactions on the carbon surface of PC–Fe3O4 surface, as reported previously.41 Therefore, this is responsible for the large irreversibility of capacity in the first cycle (Fig. 4a).45 After the charge process, the smooth layer of SEI and bulky morphology were still preserved, efficiently demonstrating the stability of the electrode. As characterized by the TEM image (Fig. 1b), the Fe3O4 nanocrystals were encapsulated within the porous carbon, and then the pulverization of Fe3O4 could be largely reduced; thus, obtaining a good cycle ability.
 | ||
| Fig. 8 SEM image of (a and b) pristine, (c and d) discharged and (e and f) charged electrode in the first cycle with different magnifications. Scale of (a, c and e) and (b, d and f) are 10 μm and 2 μm respectively. | ||
Although Na2FeP2O7 has been recently developed as a new cathode in the sodium ion battery,46–49 Examples are still lacking to apply them in a full battery versus a metal oxide, and investigate its performance. Herein, we try to introduce it into the sodium ion battery versus the anode of PC–Fe3O4. In this way, both the anode and cathode are environmental materials with a large abundance in the earth, which are critical for maintaining the sustainability in the grid storage. To date, the cathode of layered oxides of P2-type Na[Ni0.25Fe0.5Mn0.25]O2 and NASICON structured Na3V2(PO4)3 have been applied as a cathode versus Fe3O4-based anode for a full battery,19,21 but the drawbacks of the weak stability of layered oxides and high toxicity of vanadium would inevitably induce a safety problem and limit their practical applications. Considering the strong requirement of sustainability and the safety in rechargeable batteries, the cathode Na2FeP2O7 is superior than these two types of cathodes and deserves to be investigated in the sodium ion full battery, particularly versus the PC–Fe3O4, which are available to be used at the industrial level.
The typical charge–discharge curves of Na2Fe2P2O7 and sodiated PC–Fe3O4 are shown in Fig. 9. The procedures of designing the full battery are similar to those reported recently.40,44,50 Note that the large irreversible capacity of PC–Fe3O4 was compensated via the sodiation process, in which the PC–Fe3O4 electrode was sodiated by the sodium metal by contacting with sodium metal for 30 min, rather than electrochemical compensation.27 The chemical sodiation process was the same as the lithiation one. The mass ratio of the cathode and anode was controlled around 2.32/1 considering their equal total capacity in the battery.44,50 The average voltage of Na2FeP2O7 and PC–Fe3O4 were about 2.99 and 0.71 V, and the expected voltage of the battery was around 2.28 V. By analyzing the work voltage of the cathode and anode,44 the window of the work voltage for a full battery was about 1.1–4.2 V, and the calculated voltage was around 2.28 V based on the work voltage of the cathode and anode (Fig. 9).
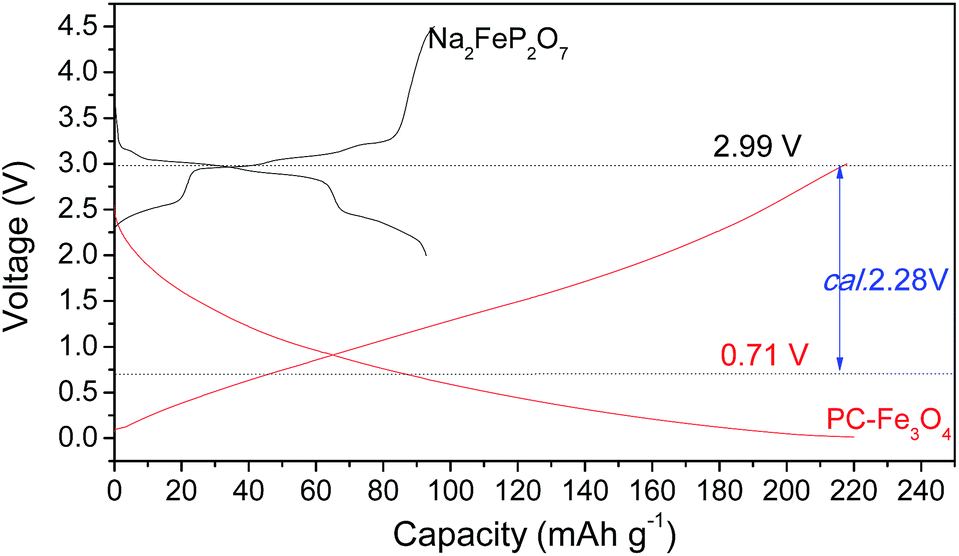 | ||
| Fig. 9 Typical charge–discharge curves of Na2FeP2O7 and lithiated PC–Fe3O4 composite. | ||
Fig. 10 shows the performance of PC–Fe3O4/Na2FeP2O7 cycled under the rate of 0.1 C. A high capacity of 93 mA h g−1 could be obtained with a work voltage of 2.28 V and an energy density of 203 W h kg−1 (which was calculated by the integral of the 1st discharge curve in Fig. 10a). In particular, the battery could be efficiently cycled over 100 cycles with the high capacity retention of 93.3% and a columbic efficiency of 98.5%, respectively. Moreover, the battery has a very excellent rate capability. For example, the capacities of 92, 89, 85, 74, 60, 46 and 32 mA h g−1 could be obtained under the rates of 0.1, 0.25, 0.5, 1, 2.5, 5 and 10 C, respectively, after 20 cycles. It is obvious that such battery has a high rate capability, which should be ascribed to the fast reaction rate of Fe3O4 nanocrystals and the fast intercalation/extraction of the sodium ion in the stable structure of Na2FeP2O7. After the high rate test, the capacity of the battery could be recovered to 90 mA h g−1 at the rate of 0.1 C, well demonstrating the stability of the electrode. More importantly, the chemical compounds required in the reversible reaction of Fe3O4 + Na2FeP2O7 ↔ NaFeP2O7 + Fe + Na2O are low-cost, abundant in the earth, environmentally friendly and rather safe. Therefore, this configuration of battery is expected to be widely used in grid energy, and it could act as energy storage for the conversion of solar and wind energy to electric power, which then can be conveniently provided for domestic uses (Fig. 11).
 | ||
| Fig. 10 Typical charge–discharge curves and capacity of PC–Fe3O4/Na2FeP2O7 under (a and b) the rate of 0.1 C and (c and d) rate test of 0.1–10 C. | ||
 | ||
| Fig. 11 Schematic of the iron-based PC–Fe3O4/Na2FeP2O7 full battery. | ||
Conclusions
A type of iron oxide-based anode of PC–Fe3O4 with a high capacity of 225 mA h g−1 at the rate of 50 mA g−1 and a stable cycle ability over 400 cycles was introduced in the sodium-ion battery for the first time. The production of PC–Fe3O4 could achieve an industrial scale towards practical application readily compared to the traditional Fe3O4 particles synthesized by the hydrothermal method. In addition, the sodium storage ability, mainly resulting from the conversion of Fe3O4 rather than porous carbon, was investigated. The stability of the electrode was further characterized by ex situ XRD and SEM. More importantly, a new concept of an elemental iron-based sodium ion battery of PC–Fe3O4/Na2FeP2O7 was presented. This is the first example to introduce an element-rich configuration in the sodium ion battery with the viewpoint of sustainability. A superior capacity of 93 mA h g−1, high capacity retention of 93.3% over 100 cycles and work voltage around 2.28 V with an energy density of 203 W h kg−1 were efficiently obtained. With the development of batteries in the energy storage field, such configuration of an iron-based sodium battery would be highly promising and sustainable owing to its low cost, high stability and safety.Acknowledgements
This work was supported by the Global Frontier R&D Program (2013-073298) on Center for Hybrid Interface Materials (HIM) funded by the Ministry of Science, ICT & Future Planning and the Human Resources Development program grant (no. 20124010203310) of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) funded by the Korean government Ministry of Trade, Industry and Energy. Industry-Academia Cooperation Innovation Fund Projects of Jiangsu Province (no. BY2011130) are gratefully acknowledged.Notes and references
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. V. Reddy, G. V. S. Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- Y. K. Sun, S. T. Myung, B. C. Park, J. Prakash, I. Belharouak and K. Amine, Nat. Mater., 2009, 8, 320–324 CrossRef CAS PubMed.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-Gonzalez and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 CAS.
- S. Goriparti, E. Miele, F. De Angelis, E. Di Fabrizio, R. P. Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS PubMed.
- J. Ming, J. B. Park and Y. K. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 2133–2136 CAS.
- X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- G. Ji, B. Ding, Y. Ma and J. Y. Lee, Energy Technol., 2013, 2, 567–572 CrossRef.
- J. Ming, Y. Wu, J. B. Park, J. K. Lee, F. Zhao and Y. K. Sun, Nanoscale, 2013, 5, 10390–10396 RSC.
- H. L. Zhu, Z. Jia, Y. C. Chen, N. Weadock, J. Y. Wan, O. Vaaland, X. G. Han, T. Li and L. B. Hu, Nano Lett., 2013, 13, 3093–3100 CrossRef CAS PubMed.
- Y. L. Cao, L. F. Xiao, M. L. Sushko, W. Wang, B. Schwenzer, J. Xiao, Z. M. Nie, L. V. Saraf, Z. G. Yang and J. Liu, Nano Lett., 2012, 12, 3783–3787 CrossRef CAS PubMed.
- D. Datta, J. W. Li and V. B. Shenoy, ACS Appl. Mater. Interfaces, 2014, 6, 1788–1795 CAS.
- L. Wu, X. H. Hu, J. F. Qian, F. Pei, F. Y. Wu, R. J. Mao, X. P. Ai, H. X. Yang and Y. L. Cao, J. Mater. Chem. A, 2013, 1, 7181–7184 CAS.
- Z. Jian, B. Zhao, P. Liu, F. Li, M. Zheng, M. Chen, Y. Shi and H. Zhou, Chem. Commun., 2014, 50, 1215–1217 RSC.
- S. Yuan, X. L. Huang, D. L. Ma, H. G. Wang, F. Z. Meng and X. B. Zhang, Adv. Mater., 2014, 26, 2273–2279 CrossRef CAS PubMed.
- H. Xiong, M. D. Slater, M. Balasubramanian, C. S. Johnson and T. Rajh, J. Phys. Chem. Lett., 2011, 2, 2560–2565 CrossRef CAS.
- K. T. Kim, G. Ali, K. Y. Chung, C. S. Yoon, H. Yashiro, Y. K. Sun, J. Lu, K. Amine and S. T. Myung, Nano Lett., 2014, 14, 416–422 CrossRef CAS PubMed.
- K. J. Harry, D. T. Hallinan, D. Y. Parkinson, A. A. MacDowell and N. P. Balsara, Nat. Mater., 2014, 13, 69–73 CrossRef CAS PubMed.
- S. M. Oh, S. T. Myung, C. S. Yoon, J. Lu, J. Hassoun, B. Scrosati, K. Amine and Y. K. Sun, Nano Lett., 2014, 14, 1620–1626 CrossRef CAS PubMed.
- S. Hariharan, K. Saravanan, V. Ramar and P. Balaya, Phys. Chem. Chem. Phys., 2013, 15, 2945–2953 RSC.
- R. R. Kumar, Y. H. Jung, K. K. Bharathi, C. H. Lim and D. K. Kim, Electrochim. Acta, 2014, 146, 503–510 CrossRef PubMed.
- N. Yabuuchi, M. Kajiyama, J. Iwatate, H. Nishikawa, S. Hitomi, R. Okuyama, R. Usui, Y. Yamada and S. Komaba, Nat. Mater., 2012, 11, 512–517 CrossRef CAS PubMed.
- D. H. Lee, J. Xu and Y. S. Meng, Phys. Chem. Chem. Phys., 2013, 15, 3304–3312 RSC.
- Z. Liu, Y. Y. Hu, M. T. Dunstan, H. Huo, X. Hao, H. Zou, G. Zhong, Y. Yang and C. P. Grey, Chem. Mater., 2014, 26, 2513–2521 CrossRef CAS.
- H. Kim, R. A. Shakoor, C. Park, S. Y. Lim, J. S. Kim, Y. N. Jo, W. Cho, K. Miyasaka, R. Kahraman, Y. Jung and J. W. Choi, Adv. Funct. Mater., 2013, 23, 1147–1155 CrossRef CAS.
- P. Barpanda, T. Ye, S. Nishimura, S. C. Chung, Y. Yamada, M. Okubo, H. S. Zhou and A. Yamada, Electrochem. Commun., 2012, 24, 116–119 CrossRef CAS PubMed.
- I. Hasa, J. Hassoun, Y. K. Sun and B. Scrosati, ChemPhysChem, 2014, 15, 2152–2155 CrossRef CAS PubMed.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef PubMed.
- J. Ming, Y. Q. Wu, G. F. Liang, J. B. Park, F. Y. Zhao and Y. K. Sun, Green Chem., 2013, 15, 2722–2726 RSC.
- H. Kim, R. A. Shakoor, C. Park, S. Y. Lim, J. S. Kim, Y. N. Jo, W. Cho, K. Miyasaka, R. Kahraman, Y. Jung and J. W. Choi, Adv. Funct. Mater., 2013, 23, 1147–1155 CrossRef CAS.
- J. Ming, H. Ming, W. J. Kwak, C. Shin, J. Zheng and Y. K. Sun, Chem. Commun., 2014, 50, 13307–13310 RSC.
- J. Ming, R. Liu, G. Liang, H. Cheng, Y. Yu and F. Zhao, J. Mater. Chem., 2014, 21, 10929–10934 RSC.
- N. C. Tombs and H. P. Rooksby, Acta Crystallogr., 1951, 4, 474–475 CrossRef CAS.
- L. B. McCusker, R. B. Von Dreele, D. E. Cox, D. Louer and P. Scardi, J. Appl. Crystallogr., 1999, 32, 36–50 CrossRef CAS.
- J. B. Moussy, J. Phys. D: Appl. Phys., 2013, 46, 143001 CrossRef.
- F. Y. Ran, Y. Tsunemaru, T. Hasegawa, Y. Takeichi, A. Harasawa, K. Yaji, S. Kim and A. Kakizaki, J. Phys. D: Appl. Phys., 2012, 45, 275002 CrossRef.
- J. Ming, Y. Wu, L. Y. Wang, Y. Yu and F. Zhao, J. Mater. Chem., 2011, 21, 17776–17782 RSC.
- H. Ming, Y. Yan, J. Ming, X. Li, Q. Zhou, H. Huang and J. Zheng, RSC Adv., 2014, 4, 12971–12976 RSC.
- J. Ming, Y. Q. Wu, G. F. Liang, J. B. Park, F. Y. Zhao and Y. K. Sun, Green Chem., 2013, 15, 2722–2726 RSC.
- H. Ming, J. Ming, S.-M. Oh, S. Tian, Q. Zhou, H. Huang, Y. K. Sun and J. Zheng, ACS Appl. Mater. Interfaces, 2014, 6(17), 15499–15508 CAS.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- A. Ponrouch, A. R. Goñi and M. R. Palacín, Electrochem. Commun., 2013, 27, 85–88 CrossRef CAS PubMed.
- K. C. Möller, H. J. Santner, W. Kern, S. Yamaguchi, J. O. Besenhard and M. Winter, J. Power Sources, 2003, 119–121, 561–566 CrossRef.
- J. Ming, W. J. Kwak, S. J. Youn, H. Ming, J. Hassoun and Y. K. Sun, Energy Technol., 2014, 2, 778–785 CrossRef CAS.
- M. R. Wagner, P. R. Raimann, A. Trifonova, K. C. Moeller, J. O. Besenhard and M. Winter, Electrochem. Solid-State Lett., 2004, 7, A201 CrossRef CAS PubMed.
- Y. H. Jung, C. H. Lim, J. H. Kim and D. K. Kim, RSC Adv., 2014, 4, 9799–9802 RSC.
- T. Honma, N. Ito, T. Togashi, A. Sato and T. Komatsu, J. Power Sources, 2013, 227, 31–34 CrossRef CAS PubMed.
- C. Y. Chen, K. Matsumoto, T. Nohira, R. Hagiwara, Y. Orikasa and Y. Uchimoto, J. Power Sources, 2014, 246, 783–787 CrossRef CAS PubMed.
- P. Barpanda, G. D. Liu, C. D. Ling, M. Tamaru, M. Avdeev, S. C. Chung, Y. Yamada and A. Yamada, Chem. Mater., 2013, 25, 3480–3487 CrossRef CAS.
- H. Ming, J. Ming, S. M. Oh, E. J. Lee, H. Huang, Q. Zhou, J. Zheng and Y. K. Sun, J. Mater. Chem. A, 2014, 2, 18938–18945 CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |