
Effect of alumina hydroxylation on glycerol hydrogenolysis to 1,2-propanediol over Cu/Al2O3: combined experiment and DFT investigation†
Pussana Hirunsit*,
Chuleeporn Luadthong and
Kajornsak Faungnawakij
National Nanotechnology Center (NANOTEC), National Science and Technology Development Agency (NSTDA), 111 Thailand Science Park, Thanon Phahonyothin, Tambon Khlong Nueng, Amphoe Khlong Luang, Pathum Thani, Thailand. E-mail: pussana@nanotec.or.th; Fax: +66 2 564 6981; Tel: +66 2 564 7100 ext. 6674
First published on 6th January 2015
Abstract
Experimental and theoretical studies were performed to investigate the glycerol hydrogenolysis to 1,2-propanediol (1,2 PD) over Cu/Al2O3 and the alumina hydration effect on catalytic activity. The experimental results show that glycerol hydrogenolysis on Cu/Al2O3 is highly active and selective to 1,2 PD, and water contained in the glycerol feedstock can decrease the catalytic reactivity. The DFT calculations demonstrate that the improved catalytic activity on Cu/Al2O3 compared to pure copper catalyst is assisted by the acidic sites (Al sites) of the alumina surface and its partial hydration. The alumina hydroxylation significantly modifies its Al site activity. The Al site and the copper site on hydroxylated alumina show similar affinity for glycerol and acetol adsorption. The Al site shows lower barrier energy of the glycerol initial O–H bond cleavage than the Cu site. However, water contained in reactants additional to water produced during the dehydration step may initially reduce the number of active Al sites, consequently, the catalytic reactivity decreases.
1. Introduction
Glycerol is one of the most important building blocks in the conversion of biomass feedstock to value-added chemicals in bio-refineries. It is an abundant by-product from biodiesel production by transesterification of vegetable oils and animal fats and the production can be close to 10 wt% of the overall biodiesel production.1–4 High purity glycerol could be obtained by a multistep purification process, and is widely used in cosmeceutical, pharmaceutical and medical applications. In addition, new catalytic processes to convert glycerol to value-added chemicals have been developed. Glycerol conversion can take place via many processes such as fermentation, oxidation, reduction, gasification, carboxylation and esterification.2,5Glycerol contains high O/C content; therefore glycerol C–O hydrogenolysis is one of the most appealing methods.1 Glycerol hydrogenolysis produces various chemical products and among all products, some are very industrially important particularly 1,3 propanediol (1,3 PD),6–12 1,2 propanediol (1,2 PD; propylene glycerol),13–21 lactic acid,22,23 1-propanol, 2-propanol and ethylene glycol (EG). The 1,3 PD is the most attractive product from glycerol conversion due to its high market price and great demand. 1,3 PD is used as a monomer of biodegradable polyester (polypropylene terephthalate, PPT) which has gained increased attention recently.1,24 The propanol and EG are not considered as the main targets of glycerol conversion because of the lower market price, poorer carbon atom efficiency and excessive hydrogen atoms consumption.1,24 1,2 PD is a non-toxic substance and is extensively used as a monomer for polyester resins which are applied in many consumer products such as paints, liquid detergent, and cosmetics.1 Currently 1,2 PD is commercially produced from propylene oxide.24 Although the cost of producing 1,2 PD from glycerol conversion is higher compared to the current method and it is a key barrier, glycerol conversion approach is a promising one for the sustainable development of our society.
A number of studies have focused on the conversion of glycerol to 1,2 PD using heterogeneous catalysts. Hydrogenolysis of glycerol into 1,2 PD proceeds over various metal catalysts such as Ru,25 Pt,26–28 Rh,29,30 Ni,31,32 Cu13,33 and supported metal on TiO2, SiO2, ZrO2 and Al2O3.19,20,34–38 The activity and selectivity of the reaction can be directed by the reaction conditions and nature of the catalysts. Pd/C showed low activity to 1,2 PD while Pt/C showed higher selectivity (82.7%) and higher yield (28.6%) which is also higher than that on Ru/C (40% selectivity and 17.5% yield).39 The noble metal catalysts often promote C–C cleavage, consequently relatively poor propanediol selectivity. Copper is known for its poor C–C cleavage activity and good for C–O bond cleavage which formally occurs during hydrogenolysis process.26,39,40
As a less expensive alternative, copper draws great attention for glycerol hydrogenolysis. Cu-based catalysts on various supports such as SiO2,13,41 Al2O3,16,25,35 ZnO–Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 and MgO–Al2O321 exhibited high selectivity (>90%) for 1,2 PD and high glycerol conversions (>75%). It has been clearly demonstrated that the influence of the support is crucial for the reaction. Nevertheless, the glycerol hydrogenolysis mechanism pathway still remains in questions. The glycerol hydrogenolysis to 1,2 PD reaction schematic is shown in Scheme 1. The selective conversion of glycerol to propanediols is known as a bi-functional reaction which requires catalysts both for dehydration and for hydrogenation functionality. On Pt/Al2O3 catalyst, it has shown that the presence of an acid–base and a metal function plays role in the dehydration of glycerol.42,43 Also, several theoretical studies using DFT calculations attempted to investigate the elementary reaction of glycerol conversion and glycerol decomposition on metallic surfaces such as Pt(111) and Rh(111).30,44–46
20 and MgO–Al2O321 exhibited high selectivity (>90%) for 1,2 PD and high glycerol conversions (>75%). It has been clearly demonstrated that the influence of the support is crucial for the reaction. Nevertheless, the glycerol hydrogenolysis mechanism pathway still remains in questions. The glycerol hydrogenolysis to 1,2 PD reaction schematic is shown in Scheme 1. The selective conversion of glycerol to propanediols is known as a bi-functional reaction which requires catalysts both for dehydration and for hydrogenation functionality. On Pt/Al2O3 catalyst, it has shown that the presence of an acid–base and a metal function plays role in the dehydration of glycerol.42,43 Also, several theoretical studies using DFT calculations attempted to investigate the elementary reaction of glycerol conversion and glycerol decomposition on metallic surfaces such as Pt(111) and Rh(111).30,44–46
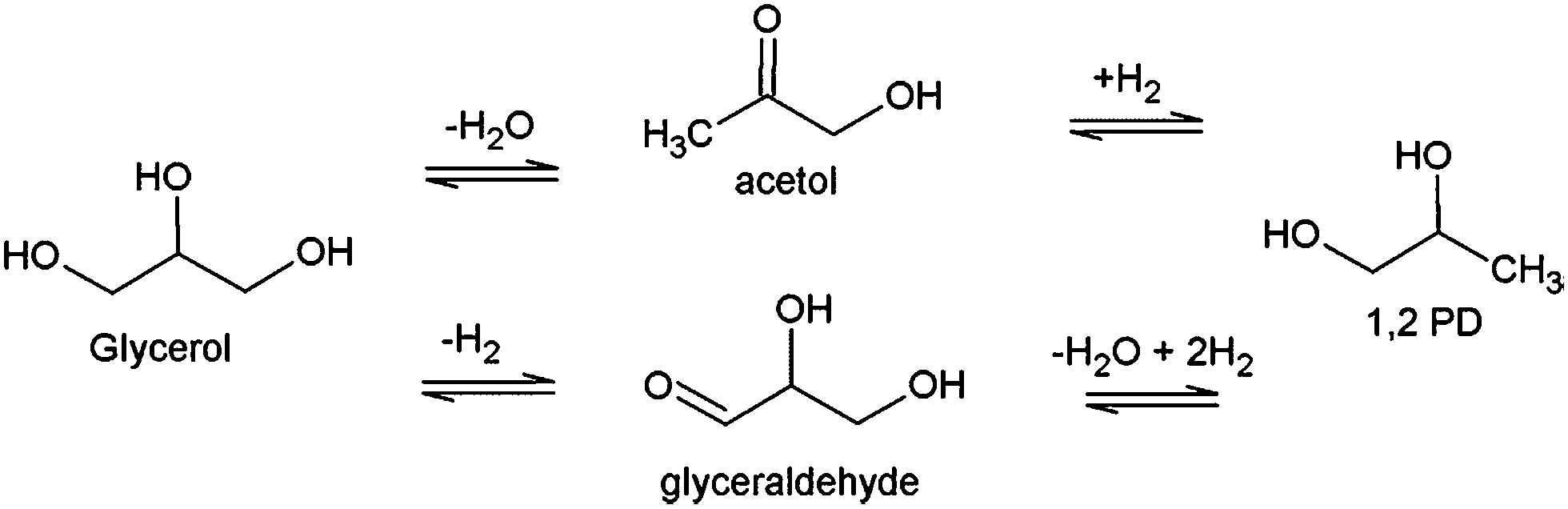 | ||
| Scheme 1 Glycerol conversion to 1,2 PD. | ||
Cu–Al catalyst system has been proposed for glycerol hydrogenolysis with high selectivity.33,34 Mizugaki et al. reported Cu nanoparticles prepared from Cu–Al hydrotalcite gave over 99% yield of 1,2 PD.47 Kwak et al. found that the CuAl2O4 prepared using sol–gel method showed glycerol conversion of 90% with 90% selectivity and 83% yield of 1,2 PD at 220 °C, 50 bar of H2 initial pressure, and 12 h of reaction time.48 However, the preparation of the spinel is generally required high calcination temperature, and would cause difficulty on scaling-up of the synthesis and the cost effectiveness. In this sense the common impregnation method of Cu on alumina is a promising way to simply prepare the catalyst with scaling-up capability. Additionally, the understanding of the copper and alumina roles in this process has been rarely reported. Under the hydrogenolysis condition, water molecules are continuously produced and possibly affect the catalytic system. The presence of water on the surface alumina was shown to facilitate the heterolytic splitting of methane and H2 dissociation.49,50 The elucidation of how the partial hydration effects on surface reactivity is important in order to understand the nature of catalyst and its key factors governing activity and selectivity of the glycerol conversion.
In this work, we conducted the experimental and theoretical study to investigate the glycerol hydrogenolysis to 1,2-propanediol (1,2 PD) over Cu/Al2O3 and to demonstrate that the alumina acidic active sites are important for the catalytic reactivity. Also, this work presents the impact on the reactivity of the acidic site on alumina support and the metallic Cu site induced by partial hydration on alumina surface.
2. Experimental details
2.1 Catalyst preparation and characterization
Alumina-supported Cu catalysts (Cu/Al2O3) were prepared by conventional impregnation method with loading of Cu at 35 wt%. The γ-Al2O3 support was commercially obtained from Sasol company, Germany. The impregnated samples were dried overnight and then calcined at 450 °C for 4 h. Unsupported copper catalysts were obtained commercially from Sigma Aldrich.The specific surface area was analyzed by a N2 sorption technique at −196 °C on the Nova 2000e, Quantachrome Instruments, Germany. Prior to the measurement, the samples were degassed at 120 °C for 1 h. The hydrogen temperature programmed reduction (H2-TPR) was conducted using a CHEMBET-Pulsar Quantachrome Instruments with a thermal conductivity detector (TCD). The catalyst (20 mg) was reduced in 5 vol% H2/Ar at a flow rate of 30 mL min−1 at a heating rate of 10 °C min−1 from 100 to 800 °C. X-ray diffraction (XRD) patterns of powder samples were collected on an X-ray diffractometer (D8 ADVANCE, Bruker, Ltd., Germany) using a Cu Kα radiation. The measurement was operated at 40 kV and 40 mA, and in step of 0.02° s−1 with a step time of 0.5 s over the range of 10° < 2θ < 80°.
2.2 Evaluation of catalytic properties
All glycerol hydrogenolysis reactions were conducted in a 130 mL stainless steel autoclave with a stirrer. Prior to each reaction test, the prepared catalyst was reduced in 10% H2/N2 at 300 °C for 3 h, typically, 10 g of glycerol (Sigma-Aldrich, 99.5%) and 1 g of a pre-reduced catalyst were loaded into the autoclave. The autoclave was pressurized with H2, (Praxair, 99.999%) to 5 MPa. Then, the reactor was heated to the desired temperature. The initial reaction pressure was observed to increase with the elevated temperature and reached its maximum when the temperature reached the desired value. After that, the pressure was found to decrease slowly with the reaction time indicating the hydrogen consumption during the reaction test. The tests were repeated twice, and the variability of data are less than 5%. After the test, the autoclave was cooled down to an ambient temperature with an ice-water bath for 20 min, followed by depressurization. Liquid-phase product was centrifuged to remove the solid catalyst powder. The chemical compositions in the liquid were analyzed by gas chromatography (GC-2010, Shimadzu) equipped with a flame ionization detector and a capillary column (DB-WAX, Agilent technologies, 30 m in length with 0.25 mm i.d. and 0.25 μm film thickness). The glycerol conversion, product yield and selectivity are calculated using the following equations:
 | (1) |
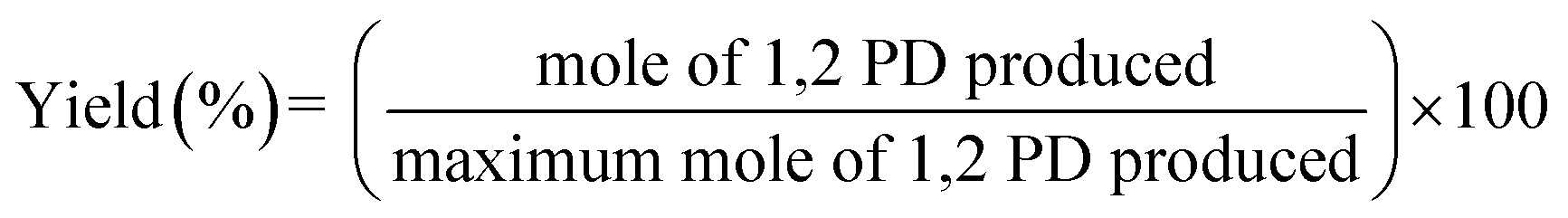 | (2) |
 | (3) |
2.3 Computational details
The slab model of Cu(111) and Cu(100) surfaces with a unit cell of 4 × 4 and 3 × 3, respectively, containing 4 layers of metal atoms and a vacuum region approximately 15 Å was applied. The two atomic layers from the bottom of the slab were fixed, while the top two layers were relaxed to their lowest energy configurations. The fixed layers were set to Cu bulk bond distances according to the optimized lattice constant that was determined from bulk calculation. The calculated Cu lattice constant is 3.63 Å. The slab model of γ-Al2O3(110) surface contains twenty four Al2O3 molecular units and ∼15 Å of the vacuum region excluding adsorbates. The (110) crystalline surface was chosen because it dominates in γ-alumina nanocrystallites (∼70–83% of total area).51,52 Eight of Al2O3 molecular units located in atomic layers from bottom of the slab were fixed and the rest were relaxed including the Cu4 cluster and adsorbates. The fixed layers were set to γ-Al2O3 bulk bond distances in which the γ-Al2O3 bulk model structure was taken from that presented by Digne et al.,51 and Krokidis et al.53 The Cu4 cluster was placed on the γ-Al2O3(110) surface initially in both planar and tetrahedral orientations. The tetrahedral Cu4 was found to be more stable on γ-Al2O3(110) surface than the planar Cu4, thus the tetrahedral Cu4/γ-Al2O3(110) model was employed. The 4-atom Cu cluster was chosen based on the fact that it is the smallest unit that can provide a three-dimensional structure (the tetrahedral structure) presenting Cu–Cu and Cu–support interaction. It should be noted that the copper particle size can affect the reactivity as well which may due to the variation of the amount of low coordinated active atoms. The supported metal-4 cluster on γ-Al2O3 surface was previously employed to study CO, C2H4 adsorption, CO2 hydrogenation, and CH4 and H2 dissociation.54–56Adsorption of water on alumina creates hydroxyls covered the surface. The relationship between the hydroxyl coverage on γ-Al2O3(110) surface and temperature has been investigated previously by Digne et al.51 Applying this relationship, the OH coverage on γ-Al2O3(110) surface corresponding to the experimental temperature in the range of 200–300 °C is approximately 5.9–11.8 OH/nm−2. The 5.9 OH/nm−2 coverage corresponding to 4H2O molecules on the γ-Al2O3(110) surface model was employed in this work. The (110) surface exhibits a great variety of surface hydroxyl groups due to many differences of local environments of the Al and O surface atoms. We performed a variety of hydroxyl group adsorption locations particularly around the copper cluster in order to bring in the effect of alumina hydroxylation on copper cluster reactivity. The bare structure of γ-Al2O3(110) and the most favorable structure of Cu4/γ-Al2O3(110) are shown in the ESI (Fig. S1†).
The density functional theory calculations, as implemented in the Vienna ab initio Simulation Program57,58 were applied. The calculations used the GGA-PBE functional59 and the Projector Augmented Wavefuction (PAW)60,61 method for representing the non-valence core electrons. For all calculations reported herein, we used a 400 eV cutoff for the kinetic energy of the plane-wave basis-set. The Methfessel–Paxton smearing62 of order 1 with a value of smearing parameter σ of 0.1 eV and the Gaussian broadening63 with a smearing width of 0.1 eV was employed for Cu(111), (100) and γ-Al2O3 surfaces, respectively. The surface Brillouin zone was sampled with a 3 × 3 × 1 Monkhorst–Pack k-point mesh64 for Cu(111), (100) and 2 × 2 × 1 for Cu4/γ-Al2O3(110). The results were checked for convergence with respect to energy cutoff and number of k-points. The convergence criterion for electronic self-consistent iteration was set to 10−7 eV and the ionic relaxation loop was limited for all forces smaller than 0.035 eV Å−1 for free atoms. The transition states (TS) structures were located using nudged elastic band (NEB)65 and dimer66 methods. The TS structures were characterized by a normal mode analysis to ensure that it features one imaginary frequency. The binding energies of the adsorbates on surfaces were calculated as
| BEabsorbate = E(adsorbate/Cu) − E(Cu) − E(adsorbate (g)) | (4) |
| BEabsorbate = E(adsorbate/Cu4/γAl2O3(110)) − E(Cu4/γAl2O3(110)) − E(adsorbate (g)) | (5) |
3. Results and discussion
3.1 Characterization of Cu catalysts
The γ-Al2O3 has a specific surface area of 201.1 m2 g−1 and pore volume of 0.550 cm3 g−1. The surface area and pore volume were lowered to 123.4 m2 g−1 and 0.306 cm3 g−1 after the Cu loading. This indicated the success deposition of copper species onto the alumina. The decrease in the surface area and pore volume by ca. 38–43% in the copper loaded sample compared to the Al2O3 corresponded to the amount of loaded Cu species along with the partial copper coverage and blockage on the surfaces and pores of Al2O3. Nonetheless, the average pore size of the catalysts remained almost constant at 7.5 nm. Note that the unsupported Cu and CuO had extremely low surface area which is less than 2 m2 g−1. As shown in Fig. 1, the XRD patterns of the fresh catalysts reveal the CuO phase, while the Al2O3 showed low crystallinity or amorphous structure. After the reduction, CuO was transformed to metallic copper phase, and no oxide phase was detected over all samples. The TEM (transmission electron microscopy) images (Fig. S2 in ESI†) shows bare Cu is a dense particle with sphere-like shape, and its size is in the range of 20–40 nm. For Cu/Al2O3, the particle of copper is in the range of 2–10 nm. | ||
| Fig. 1 XRD patterns of the catalysts (a) CuO and (b) Cu/Al2O3 before and after reduction: CuO (♦), Cu0 (□), Al2O3 (▽). | ||
Additionally, NH3-TPD over Al2O3 and Cu/Al2O3 (Fig. S3 in ESI†) suggested the presence of acid site which is expected to contribute to the surface reaction in hydrogenolysis which is discussed later in Section 3.3. The acid function was mainly from Al2O3 surface since the NH3 desorption curves appearing from 150 to 700 °C from the catalysts before and after copper loading were nearly comparable.
3.2 Catalytic properties of hydrogenolysis of glycerol on Cu catalysts
The catalytic activity of the catalysts is shown in Table 1. The bare copper catalyst gave a low conversion of 15% and low 1,2 PD yield of 4.8%. Acetol as an intermediate and ethylene glycol as a side-product along with other unknown species were also observed. The pure Al2O3 showed a low conversion of 7%, and yielded traces of products. The improved conversion of 61% with 1,2 PD yield of 56.9% and 1,2 PD selectivity of 93.3% was achieved over Cu/Al2O3. Clearly, the alumina support played a vital role on catalysis of Cu/Al2O3. Without the alumina support, the selectivity to 1,2 PD was low at 33%, which suggests that the reaction rates to byproducts become larger or the intermediates could not undergo the complete conversion to 1,2 PD. However, the bare alumina could not catalyze the glycerol hydrogenolysis itself. Therefore, the synergic effect of Cu and alumina on Cu/Al2O3 is a major contribution to effectively drive the reaction. Note that 1,3 PD was not observed for all experiments.| Catalysts | Conversion | %Yield | %Selectivity | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1,2 PD | Acetol | Ethylene glycol | Other intermediates/unknowns | 1,2 PD | Acetol | Ethylene glycol | Other intermediates/unknowns | ||
| a Reaction conditions: temperature of 220 °C, initial pressure of 5 MPa, time of 6 h. | |||||||||
| Cu0 | 15 | 4.8 | 0.1 | 0.1 | 10.0 | 32.0 | 0.7 | 0.7 | 66.6 |
| Al2O3 | 7 | 0.1 | 0 | 0 | 6.9 | 1.4 | 0 | 0 | 98.6 |
| Cu/Al2O3 | 61 | 56.9 | 0.4 | 1.1 | 2.6 | 93.3 | 0.7 | 1.8 | 4.3 |
During the hydrogenolysis, water is produced as a co-product and possibly has an impact on catalytic activity. Therefore, it is of interest to study the influence of water presence on the catalytic behavior. As shown in Fig. 2, the addition of water to the reaction resulted in lower conversion and lower 1,2 PD yield at the reaction time of 1 hour compared to that in the case of initial water-free condition. The conversion and 1,2 PD yield at both conditions become comparable with the prolonged reaction time. This suggested that the initial presence of water decreases the reactivity for some periods of time, consequently, the total production reduces. Water molecules may affect, for example, the surface active sites, surface chemistry and the stability of intermediate species which is demonstrated in the theoretical study. The identical yield of the product at 6 h infers that the major reaction pathways would be the same, while the overall production rate reduces when the reactants contain water.
 | ||
| Fig. 2 Effect of water addition on the catalyst performance in glycerol hydrogenolysis. Reaction conditions: temperature of 220 °C, initial pressure of 5 MPa, time of 1 and 6 h. | ||
Furthermore, the spent catalysts without any post treatment were analyzed by XRD as shown in Fig. 3. The results suggested that the crystalline phase of the catalyst remained stable before and after the reaction tests in both water-free and water-added initial conditions, and the metallic copper was considered the active site for the hydrogenolysis.
 | ||
| Fig. 3 XRD patterns of (a) Cu/Al2O3 catalyst after the reaction (10 wt% water), (b) Cu/Al2O3 catalyst after the reaction (no initial water), and (c) Cu/Al2O3 catalyst after reducing at 300 °C: metallic Cu (□), Al2O3 (▽). | ||
3.3 Calculation results
Glycerol and acetol interaction with Cu(111), Cu(100) surfaces, and Cu4/γ-Al2O3(110) surfaces with and without hydroxylation were investigated using DFT calculations. Acetol is a key intermediate which is found in glycerol hydrogenolysis. Acetol is a primary product upon glycerol dehydration. Then, 1,2 PD is produced via acetol hydrogenation. The glycerol transformation into 1,2 PD is generally a dehydration coupled to a hydrogenation; however, the mechanism is still unclear.30,44,67,68 The first step of either glycerol dehydration or dehydrogenation is still debatable. Upon dehydration step, the alumina surface could be hydroxylated. The hydration of the γ-Al2O3 surface is important as it modifies the number of active acidic sites and determines the nature of γ-Al2O3 catalytic properties.49,50 The hydroxyl groups adsorbed on γ-Al2O3 are the products of water dissociation which effectively takes place on alumina surface.49,51,69 We performed DFT calculations to investigate the influence of the alumina support and its partial hydration on the stability of the key species (i.e. glycerol and acetol) and the early stage of glycerol O–H bond scission.Table 2 shows adsorption energies of the most stable glycerol and acetol on Cu(111), Cu(100) and Cu4/γ-Al2O3(110) with and without hydroxylation on alumina surface in which a number of possible adsorption sites interacting with Cu sites and Al sites were investigated. The Cu4 cluster size is smaller than the Cu particles reported from the experiment as seen in TEM analysis. It should be noted that the Cu cluster size and shape has influence on catalytic reactivity, however, in this work we would like to demonstrate the different activity at Al and Cu site and the important role of alumina surface and its partial hydroxylation. Also the Cu4 cluster on alumina support model have been applied in several studies and yielded agreeable results on C2H4 adsorption, CO2 hydrogenation, and CH4 and H2 dissociation.54–56 The most favorable structures corresponding to the structures discussed in Table 2 are shown in Fig. 4 and 5. Cu(111) and Cu(100) interacts to glycerol and acetol relatively weak which may results in very low glycerol conversion and very low 1,2 PD yield observed experimentally on pure Cu compared to that on Cu4/γ-Al2O3. Glycerol and acetol binds to Cu4/γ-Al2O3 significantly stronger than pure Cu. The calculations suggest that the acidic sites (Al sites) on alumina support are more favorable for glycerol and acetol adsorption than the Cu sites. When the alumina surface is not partly hydroxylated, the spontaneous dissociation of glycerol O–H occurs at Al site and we cannot found a stable non-dissociated structure of glycerol adsorption on alumina surface. The highly favorable of the dissociated glycerol adsorption on alumina surface (non-hydroxylated) may cause the particularly low conversion observed experimentally on pure alumina where the further elementary step cannot proceed. Generally, the dehydration steps are associated to catalysis via an acidic function of the support, whereas the hydrogenation or dehydrogenation reactions need a metallic function. The acidic degree of the Al site is reduced due to the surface hydroxylation. Thus, the activity toward glycerol dissociation decreases and we do not found the spontaneous glycerol O–H dissociation on the hydroxylated alumina surface.
| Glycerol adsorption (eV) | Acetol adsorption (eV) | |||
|---|---|---|---|---|
| Cu site | Al site | Cu site | Al site | |
| Cu(111) | −0.15 | — | −0.36 | — |
| Cu(100) | −0.27 | — | −0.44 | — |
| Cu/non-hydroxylated γ-Al2O3(110) | −0.85 | Spontaneous O–H breaking | −1.05 | −5.02 |
| Cu/hydroxylated γ-Al2O3(110) | −0.90 | −1.02 | −1.15 | −1.45 |
 | ||
| Fig. 4 The most stable configurations of glycerol adsorption on (a) Cu(111), (b) Cu(100), (c) Cu4/non-hydroxylated γ-Al2O3(110) and (d) Cu4/hydroxylated γ-Al2O3(110). Yellow atom is H from glycerol O–H breaking, green atoms are O from hydroxylation, blue atoms are O of glycerol, and grey atoms are C. | ||
 | ||
| Fig. 5 The most stable configuration of acetol adsorption on (a) Cu(111), (b) Cu(100), (c) Cu4/non-hydroxylated γ-Al2O3(110) and (d) Cu4/hydroxylated γ-Al2O3(110). Green atoms are O from hydroxylation, blue atoms are O of acetol, and grey atoms are C. | ||
Additionally, the hydroxylation significantly weakens the interaction between glycerol and γ-Al2O3 surface (Table 2) which agrees with the experimental study by Copeland et al.70 They investigated the interactions between polyols with two and three carbon atoms (i.e. glycerol, 1,2 PD, 1,3 PD and ethylene glycol) and γ-Al2O3 using transmission IR and nuclear magnetic resonance (NMR) spectroscopic techniques and found that the competitive adsorption between coadsorbed water and the polyols limits their uptake from aqueous solutions on γ-Al2O3.70 Nevertheless, the most stable structure of glycerol adsorption on hydrated γ-Al2O3 surface given by DFT calculations in this work is different from that reported by Copeland et al.70 Their DFT results showed that hydrogen atom of one of the terminated OH groups dissociates, the two oxygen atoms of the glycerol terminated OH group bind with two coordinately unsaturated Al atoms and the secondary OH group forms a hydrogen bond to the surface. This work found that upon hydroxylation glycerol does not dissociate and binds to Al with the secondary OH group and the other OH groups tend to form hydrogen bonds with surface atoms (Fig. 4(d) Al site). Although the OH coverage in Copeland et al.70 and this work is equivalent, the uncoordinated unsaturated Al-atom sites on γ-Al2O3 surface are not similar due to the copper cluster presence in this work. The substantial difference in surface geometry may cause the disagreement about the most favorable adsorption configuration.
Furthermore, it was previously reported that on Pt(111) the C–C scission is likely to be a very slow process, with rapid C–H or O–H bond scission dominating for the early stage of glycerol decomposition.46 Also, the barrier energy of the C–H and O–H bond scission was shown to be comparable with slightly higher for the O–H bond cleavage.46 Here, we report the energy barrier for the O–H bond scission to investigate the active site for the early stage of glycerol decomposition. The calculated barrier energy of the terminated O–H bonding cleavage is 1.29 eV on Cu(111) and 0.84 eV on Cu(100). The calculated barrier energy of the central O–H bonding cleavage is 1.01 eV on Cu(111) and 0.87 eV on Cu(100). The activation energy of glycerol O–H dissociation on hydroxylated alumina surface was calculated to be 0.65 eV while that at Cu site are 1.47 and 1.43 eV for terminated O–H and central O–H, respectively. The transition state structures are shown in the ESI (Fig. S4 and S5†). The calculations indicate that the glycerol O–H dissociation on Cu(111) and on Cu(100) surfaces is more kinetically limited than on Cu4/Al2O3. The acidic Al site is more active than the Cu site for glycerol O–H dissociation on Cu4/Al2O3. The glycerol hydrogenolysis mechanism consists of many elementary steps and it is unclear which step is the rate-limiting one. The calculations do not include the entire pathway; however, it can demonstrate the different activity at Al and Cu site and the important role of alumina surface and its partial hydroxylation.
Similar to glycerol adsorption, acetol adsorbs significantly weaker on Cu(111) and Cu(100) than that on Cu4/γ-Al2O3 surface (Table 2). Also, acetol adsorption is more favorable at Al site than the Cu site. The hydroxylation on alumina support shows slight effect on acetol interaction with the Cu site. However, the hydroxylation shows a significant effect on acetol interaction at the Al site that the hydroxylation considerably decreases acetol adsorption energy at the Al site. The hydrogenation process has been proposed to require the synergic effect between the metallic site and the acidic site on oxide support.1,39,68,71 The metal site is required for H2 dissociation and hydrogen atom reacts to an adsorbed intermediate on the support. Therefore, the substantial strong acetol adsorption on non-hydroxylated alumina surface may significantly prevent acetol to further react. This suggests that the hydroxylation of alumina possibly facilitate acetol hydrogenation process. Also, the hydroxylation might prohibit a too high quantity of acidic sites which can promote C–C bond cleavages via an acidic cracking mechanism under H2 atmosphere and the carbon chain is not maintained resulting in relatively high yield of EG (≤C2) products. Nevertheless, water in reactants additional from those produced by dehydration step reduces the initial reactivity observed experimentally may be ascribed to the decrease in number of acidic sites on alumina and initially more activated side reaction of water dissociation. Until the hydroxylation coverage reaches its equilibrium at the reaction temperature, the additional amount of water does not alter the catalysis.
4. Conclusions
The combined experimental and theoretical study of glycerol hydrogenolysis to 1,2 PD over Cu/Al2O3 and the alumina hydration effect on catalytic activity is performed. The experimental results showed that Cu/Al2O3 supported catalyst is highly active and selective as compared with single component of alumina and copper. The theoretical study reveals that the partial hydrations on the alumina support has a significant impact on intermediates stability and the reactivity of the glycerol initial O–H bond cleavage. The alumina support facilitates copper to be more active toward interacting with glycerol and acetol intermediate species which results in the improved catalytic activity compared to the pure copper catalyst. The role of alumina surface and its hydroxylation is shown to be crucial for the reaction. The Al site (acidic site) could be as active as the Cu site toward glycerol and acetol adsorption when the alumina surface is partially hydroxylated. Nevertheless, water contained in reactants additional from those produced upon dehydration step could initially reduce the number of Al active site and is found to decrease the reactivity at some periods of time.Acknowledgements
The authors acknowledge financial supports from the National Nanotechnology Center (NANOTEC), NSTDA, and the Thailand Research Fund (TRF) to K.F and P.H. and the computing resource from National e-Science Infrastructure Consortium by the National Electronics and Computer Technology Center (NECTEC). The authors also thank Dr Chalita Ratanatawanate for the TEM analysis assistance.Notes and references
- Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2011, 1, 179–190 CAS.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed.
- C. H. Zhou, H. Zhao, D. S. Tong, L. M. Wu and W. H. Yu, Catal. Rev.–Sci. Eng., 2013, 55, 369–453 CAS.
- C.-W. Chiu, M. A. Dasari, W. R. Sutterlin and G. J. Suppes, Ind. Eng. Chem. Res., 2005, 45, 791–795 CrossRef.
- C.-H. Zhou, J. N. Beltramini, Y.-X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- L. Gong, Y. Lu, Y. Ding, R. Lin, J. Li, W. Dong, T. Wang and W. Chen, Appl. Catal., A, 2010, 390, 119–126 CrossRef CAS PubMed.
- Y. Amada, Y. Shinmi, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2011, 105, 117–127 CrossRef CAS PubMed.
- T. Kurosaka, H. Maruyama, I. Naribayashi and Y. Sasaki, Catal. Commun., 2008, 9, 1360–1363 CrossRef CAS PubMed.
- Y. Nakagawa, Y. Shinmi, S. Koso and K. Tomishige, J. Catal., 2010, 272, 191–194 CrossRef CAS PubMed.
- Y. Nakagawa, X. Ning, Y. Amada and K. Tomishige, Appl. Catal., A, 2012, 433–434, 128–134 CrossRef CAS PubMed.
- S. Zhu, X. Gao, Y. Zhu, Y. Zhu, X. Xiang, C. Hu and Y. Li, Appl. Catal., B, 2013, 140–141, 60–67 CrossRef CAS PubMed.
- R. Arundhathi, T. Mizugaki, T. Mitsudome, K. Jitsukawa and K. Kaneda, ChemSusChem, 2013, 6, 1345–1347 CrossRef CAS PubMed.
- Z. Huang, F. Cui, H. Kang, J. Chen, X. Zhang and C. Xia, Chem. Mater., 2008, 20, 5090–5099 CrossRef CAS.
- C.-W. Chiu, A. Tekeei, W. R. Sutterlin, J. M. Ronco and G. J. Suppes, AIChE J., 2008, 54, 2456–2463 CrossRef CAS.
- C.-W. Chiu, A. Tekeei, J. M. Ronco, M.-L. Banks and G. J. Suppes, Ind. Eng. Chem. Res., 2008, 47, 6878–6884 CrossRef CAS.
- M. Akiyama, S. Sato, R. Takahashi, K. Inui and M. Yokota, Appl. Catal., A, 2009, 371, 60–66 CrossRef CAS PubMed.
- S. Sato, M. Akiyama, K. Inui and M. Yokota, Chem. Lett., 2009, 38, 560–561 CrossRef CAS.
- A. Bienholz, H. Hofmann and P. Claus, Appl. Catal., A, 2011, 391, 153–157 CrossRef CAS PubMed.
- S. Zhu, X. Gao, Y. Zhu, Y. Zhu, H. Zheng and Y. Li, J. Catal., 2013, 303, 70–79 CrossRef CAS PubMed.
- Y. Feng, H. Yin, A. Wang, L. Shen, L. Yu and T. Jiang, Chem. Eng. J., 2011, 168, 403–412 CrossRef CAS PubMed.
- Z. Yuan, L. Wang, J. Wang, S. Xia, P. Chen, Z. Hou and X. Zheng, Appl. Catal., B, 2011, 101, 431–440 CrossRef CAS PubMed.
- R. K. P. Purushothaman, J. van Haveren, D. S. van Es, I. Melián-Cabrera, J. D. Meeldijk and H. J. Heeres, Appl. Catal., B, 2014, 147, 92–100 CrossRef CAS PubMed.
- D. Roy, B. Subramaniam and R. V. Chaudhari, ACS Catal., 2011, 1, 548–551 CrossRef CAS.
- D. T. Johnson and K. A. Taconi, Environ. Prog., 2007, 26, 338–348 CrossRef CAS.
- M. Balaraju, V. Rekha, P. S. S. Prasad, B. L. A. P. Devi, R. B. N. Prasad and N. Lingaiah, Appl. Catal., A, 2009, 354, 82–87 CrossRef CAS PubMed.
- C. Montassier, J. C. Ménézo, L. C. Hoang, C. Renaud and J. Barbier, J. Mol. Catal., 1991, 70, 99–110 CrossRef CAS.
- E. P. Maris and R. J. Davis, J. Catal., 2007, 249, 328–337 CrossRef CAS PubMed.
- C. Deng, X. Duan, J. Zhou, D. Chen, X. Zhou and W. Yuan, Catal. Today, 2014, 234, 208–214 CrossRef CAS PubMed.
- I. Furikado, T. Miyazawa, S. Koso, A. Shimao, K. Kunimori and K. Tomishige, Green Chem., 2007, 9, 582–588 RSC.
- F. Auneau, C. Michel, F. Delbecq, C. Pinel and P. Sautet, Chem.–Eur. J., 2011, 17, 14288–14299 CrossRef CAS PubMed.
- A. Perosa and P. Tundo, Ind. Eng. Chem. Res., 2005, 44, 8535–8537 CrossRef CAS.
- L. Huang, Y.-L. Zhu, H.-Y. Zheng, Y.-W. Li and Z.-Y. Zeng, J. Chem. Technol. Biotechnol., 2008, 83, 1670–1675 CrossRef CAS.
- Z. Wu, Y. Mao, M. Song, X. Yin and M. Zhang, Catal. Commun., 2013, 32, 52–57 CrossRef CAS PubMed.
- S. Wang and H. Liu, Chin. J. Catal., 2014, 35, 631–643 CrossRef CAS.
- D. Sun, Y. Yamada and S. Sato, Appl. Catal., A, 2014, 475, 63–68 CrossRef CAS PubMed.
- S. N. Delgado, L. Vivier and C. Especel, Catal. Commun., 2014, 43, 107–111 CrossRef CAS PubMed.
- D. Durán-Martín, M. Ojeda, M. L. Granados, J. L. G. Fierro and R. Mariscal, Catal. Today, 2013, 210, 98–105 CrossRef PubMed.
- M. Balaraju, V. Rekha, B. L. A. P. Devi, R. B. N. Prasad, P. S. S. Prasad and N. Lingaiah, Appl. Catal., A, 2010, 384, 107–114 CrossRef CAS PubMed.
- M. A. Dasari, P.-P. Kiatsimkul, W. R. Sutterlin and G. J. Suppes, Appl. Catal., A, 2005, 281, 225–231 CrossRef CAS PubMed.
- J. Chaminand, L. a. Djakovitch, P. Gallezot, P. Marion, C. Pinel and C. Rosier, Green Chem., 2004, 6, 359–361 RSC.
- Z. Huang, F. Cui, J. Xue, J. Zuo, J. Chen and C. Xia, Catal. Today, 2012, 183, 42–51 CrossRef CAS PubMed.
- A. Wawrzetz, B. Peng, A. Hrabar, A. Jentys, A. A. Lemonidou and J. A. Lercher, J. Catal., 2010, 269, 411–420 CrossRef CAS PubMed.
- I. Gandarias, P. L. Arias, J. Requies, M. B. Güemez and J. L. G. Fierro, Appl. Catal., B, 2010, 97, 248–256 CrossRef CAS PubMed.
- D. Coll, F. Delbecq, Y. Aray and P. Sautet, Phys. Chem. Chem. Phys., 2011, 13, 1448–1456 RSC.
- B. Liu and J. Greeley, Phys. Chem. Chem. Phys., 2013, 15, 6475–6485 RSC.
- B. Liu and J. Greeley, J. Phys. Chem. C, 2011, 115, 19702–19709 CAS.
- T. Mizugaki, R. Arundhathi, T. Mitsudome, K. Jitsukawa and K. Kaneda, Chem. Lett., 2013, 42, 729–731 CrossRef CAS.
- B. K. Kwak, D. S. Park, Y. S. Yun and J. Yi, Catal. Commun., 2012, 24, 90–95 CrossRef CAS PubMed.
- R. Wischert, P. Laurent, C. Copéret, F. Delbecq and P. Sautet, J. Am. Chem. Soc., 2012, 134, 14430–14449 CrossRef CAS PubMed.
- J. Joubert, A. Salameh, V. Krakoviack, F. Delbecq, P. Sautet, C. Copéret and J. M. Basset, J. Phys. Chem. B, 2006, 110, 23944–23950 CrossRef CAS PubMed.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2004, 226, 54–68 CrossRef CAS PubMed.
- P. Nortier, P. Fourre, A. B. M. Saad, O. Saur and J. C. Lavalley, Appl. Catal., 1990, 61, 141–160 CrossRef CAS.
- X. Krokidis, P. Raybaud, A.-E. Gobichon, B. Rebours, P. Euzen and H. Toulhoat, J. Phys. Chem. B, 2001, 105, 5121–5130 CrossRef CAS.
- M. C. Valero, P. Raybaud and P. Sautet, J. Catal., 2007, 247, 339–355 CrossRef CAS PubMed.
- J. Li, E. Croiset and L. Ricardez-Sandoval, J. Phys. Chem. C, 2013, 117, 16907–16920 CAS.
- R. Zhang, B. Wang, H. Liu and L. Ling, J. Phys. Chem. C, 2011, 115, 19811–19818 CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS.
- C. L. Fu and K. M. Ho, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 5480–5486 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- G. Henkelman and H. Jonsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS PubMed.
- G. Henkelman and H. Jonsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS PubMed.
- J. Feng, H. Fu, J. Wang, R. Li, H. Chen and X. Li, Catal. Commun., 2008, 9, 1458–1464 CrossRef CAS PubMed.
- T. Mizugaki, T. Yamakawa, R. Arundhathi, T. Mitsudome, K. Jitsukawa and K. Kaneda, Chem. Lett., 2012, 41, 1720–1722 CrossRef.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2002, 211, 1–5 CrossRef CAS.
- J. R. Copeland, X.-R. Shi, D. S. Sholl and C. Sievers, Langmuir, 2012, 29, 581–593 CrossRef PubMed.
- P. Hirunsit, K.-i. Shimizu, R. Fukuda, S. Namuangruk, Y. Morikawa and M. Ehara, J. Phys. Chem. C, 2014, 118, 7996–8006 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The surface models for DFT calculations, the transition structures, the TEM images and NH3-TPD profiles. See DOI: 10.1039/c4ra14698k |
| This journal is © The Royal Society of Chemistry 2015 |