
The electrocatalytic activity of IrO2–Ta2O5 anode materials and electrolyzed oxidizing water preparation and sterilization effect†
Abstract
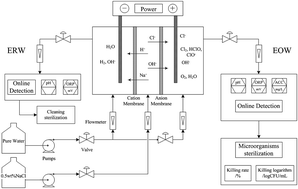
Ti/IrO2–Ta2O5 anode electrocatalysts with different contents and preparation temperatures were prepared by thermal decomposition in this work. The crystallite characterization and morphology were examined via XRD and SEM. The electrochemical properties were examined via cyclic voltammetry (CV) in 0.5 M H2SO4 and linear sweep voltammetry (LSV) in saturated sodium chloride. Through the study of different series of Ti/IrO2–Ta2O5 anodes, we find that the preparation conditions have a great impact on the electrode catalytic activity. Experimental results indicate that the electrochemically active surface area is determined by the content and morphology of the anode coating. When the IrO2 content and the preparation temperature are 70% and 500 °C, the surface of the electrode is aggregated with segregated crystallite flower-like particles, which brings about the best electrode catalytic activity. The current density in the chlorine evolution reaction of IrO2–Ta2O5 (70% and 500 °C) is 0.4 A cm−2 in saturated sodium chloride. The properties and sterilization effect of EO water are closely related to the electrode catalytic activity. The higher the current density is in chlorine evolution, the higher the available chlorine and HClO content. When the IrO2 content is 70% and the preparation temperature is 500 °C, the maximum values of the killing logarithm value and killing rate are 3.01–3.05 and 99.9023–99.9109%, respectively. In addition, when the Ti substrate undergoes 40 minutes of activation treatment, the Ti/IrO2–Ta2O5 anodes have the highest stability.
 Please wait while we load your content...
Please wait while we load your content...

