
Colorimetric and fluorogenic signalling of fluoride ions by diketopyrrolopyrrole-based chemosensor†
Xiaofeng Yang,
Gege Zhang,
Yexin Li,
Zheng Liu,
Xiaoqian Gong,
Bin Gao,
Guangyou Zhang,
Yu Cui* and
Guoxin Sun*
School of Chemistry and Chemical Engineering, University of Jinan, Jinan 250022, China. E-mail: chm_cuiy@ujn.edu.cn; chm_sungx@ujn.edu.cn; Tel: +86-53182765407
First published on 18th February 2015
Abstract
A new colorimetric and on–off fluorescence sensor based on diketopyrrolopyrrole (DPP) was synthesized for detection of fluoride anion. The sensor displayed a rapid responsivity and high selectivity and sensitivity towards F− over other anions, and it also exhibited a very low detection limit of 4.07 × 10−8 M. The signaling process was confirmed by UV-vis, fluorescence measurements as well as 1H and 19F NMR spectroscopy. The experimental results revealed that the formation of anionic species from deprotonation of the hydrazone N–H moiety by fluoride ion was responsible for the spectral changes. Density function theory calculations were conducted to rationalize the optical response of the sensor.
Introduction
Fluoride was a common ingredient in anesthetics, hypnotics, psychiatric drugs, rat and cockroach poisons, and military nerve gases, and it was also a contaminant in drinking water.1–3 Excess fluoride exposure might cause collagen breakdown, bone disorder and thyroid activity.4,5 In addition, with the development of industry and the increase in the number and the scale of enterprises on fluorine products, the contamination of fluorine became more serious. Thus, detection of fluoride with simple prepared sensor and minimal instrumental assistance was desirable towards practical applications.6In current study, the development of fluorescent sensors for anions sensing was attractive due to their simplicity, high degree of specificity and low detection limit.7–10 The reasonable construction of a fluorescent chemosensor usually involved two integrated components. One was a fluorescent signaling unite and another was a receptor for target to be detected. Various fluorescence and colorimetric chemosensors for fluoride-ion detection had been developed based on urea or thiourea, amide, phenol cationic borane, pyrrole based macrocycles, boron-dipyrromethenes (BODIPYs), and so forth.11
Diketopyrrolopyrrole (DPP) pigments were discovered in the 1980s and were mainly used as low-cost pigments for application with high-quality demands.12,13 DPPs were also applied in biophotonics and optoelectronics14 for preparation of low bandgap conjugated polymers15–18 or copolymers used in semiconductors.19 Recently, many research groups reported progress in the development of DPP-containing fluorescent sensors, and they investigated their applications in selective detection of thiols, Zn2+, cyanide, pH, DNA and CO2.20–26 However, reports on the F− detection using DPP derivatives were very few.27–31
A hydrazone (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NH–) contained hydrogen donor moiety of NH, which had the tendency for binding of anions. To the best of our knowledge, a few most of the hydrazone based receptors reported in literature were colorimetric,32–35 and colorimetric and fluorescent fluoride sensors based on hydrazone were very few.36–38 Recently, we reported DPP-based chemosensors for detection of fluoride ion.39,40 As a continuation of this research program, the present manuscript demonstrated the synthesis of a new DPP derivative 1 based on benzothiazolehydrazone group for the optical detection of fluoride ions.
N–NH–) contained hydrogen donor moiety of NH, which had the tendency for binding of anions. To the best of our knowledge, a few most of the hydrazone based receptors reported in literature were colorimetric,32–35 and colorimetric and fluorescent fluoride sensors based on hydrazone were very few.36–38 Recently, we reported DPP-based chemosensors for detection of fluoride ion.39,40 As a continuation of this research program, the present manuscript demonstrated the synthesis of a new DPP derivative 1 based on benzothiazolehydrazone group for the optical detection of fluoride ions.
Experimental section
Reagents and chemicals
2-Hydrazinobenzothiazole and all of tetrabutylammonium (TBA+) salts were purchased from Aladdin Shanghai Reagent Company. DMSO was distilled in the presence of CaH2 under reduced pressure before use. Other reagents were used without further purification. Reactions were monitored by TLC. Flash chromatography separations were carried out using silica gel (200–300 mesh).Measurements
1H NMR, 1H–1H COSY, 13C NMR, 1H–13C HSQC and 19F NMR spectra were collected on a Bruker Avance II 400 MHz spectrometer. UV-vis spectra were recorded on a Shimadzu 3100 spectrometer. Fluorescence measurements were carried out using an Edinburgh Instruments Ltd-FLS920 fluorescence spectrophotometer.General method
All UV-vis and fluorescence titration experiments were carried out at room temperature. To the 1 × 10−5 M dry DMSO solution of the receptor, the varying equivalents of the anions were added separately and spectra were recorded. Titration plots were generated by using Origin (Microcal software). The 1H NMR was carried out in DMSO-d6 using TMS as an internal reference standard. To the 4.7 × 10−3 M solution of sensor 1 in DMSO-d6 the varying equivalents of TBA+F− were added and the 1H NMR and 19F NMR spectra recorded after each addition.Synthesis
Compound 4 was synthesized according to the reported method.41 Compound 4 (280 mg, 0.65 mmol), K2CO3 (160 mg, 1.39 mmol) and 40 mL DMF were added into a three-necked flask and heated to 120 °C for 30 min. 1-Chloro-2-(2-(2-methoxyethoxy)ethoxy)ethane (876 mg, 4.8 mmol) in 10 mL DMF was then added slowly. The mixture was kept at 120 °C for 6 h. Then water was poured in to quench the reaction, and the mixture was extracted with ethyl acetate. The organic layer was dried over Na2SO4. After filtration, the solvent was removed under reduced pressure, and the crude was purified by column chromatography (silica gel; ethyl acetate/petroleum ether, 10/1, v/v) to give compound 3 (188 mg, 40%). 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.98 (d, J = 8.4 Hz, 4H), 7.61 (d, J = 8.0 Hz, 4H), 5.88 (s, 2H), 4.14–4.03 (m, 8H), 3.91 (t, J = 5.6 Hz, 4H), 3.71 (t, J = 5.6 Hz, 4H), 3.57–3.54 (m, 12H), 3.49–3.47 (m, 4H), 3.33 (s, 6H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 162.80, 148.65, 129.34, 128.58, 126.80, 109.76, 103.01, 71.85, 70.51, 70.44, 68.76, 65.30, 58.93, 42.04; element analysis for C38H48N2O12 (%): C 62.90, H 6.61, N 3.92, calculated C 62.97, H 6.68, N 3.87 (Fig. S1 and S2†).A mixture of compound 3 (159 mg, 0.22 mmol), THF (10 mL), and HCl (2 M, 5 mL) was stirred for 2 h at 60 °C. After being cooled to room temperature, DCM (40 mL) was added, and the mixture was washed with water and dried over Na2SO4. After filtration, the solvent was removed under reduced pressure, and the crude product was purified by column chromatography (silica gel; ethyl acetate/petroleum ether, 10/1, v/v) to give compound 2 (105 mg, 75%). 1H NMR (CDCl3, 400 MHz) δ (ppm) 10.09 (s, 2H), 8.20 (d, J = 8.4 Hz, 4H), 8.03 (d, J = 8.4 Hz, 4H), 3.93 (t, J = 5.2 Hz, 4H), 3.75 (t, J = 5.6 Hz, 4H), 3.58–3.55 (m, 12H), 3.50–3.48 (m, 4H), 3.34 (s, 6H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 191.37, 162.59, 148.40, 137.59, 133.19, 130.07, 129.88, 110.80, 71.85, 70.55, 70.50, 70.47, 68.77, 59.00, 42.50; element analysis for C34H40N2O10 (%): C 64.20, H 6.31, N 4.43, calculated C 64.14, H 6.33, N 4.40 (Fig. S3 and S4†).
To 20 mL of compound 2 (318 mg, 0.5 mmol) in ethanol, was added 5 mL of 2-hydrazinobenzothiazole (182 mg, 1.1 mmol) in ethanol dropwise. After the reaction solution was refluxed for 3 h, the rust red solid was precipitated, collected, and wash with ethanol to afford compound 1 (302 mg, 65%). 1H NMR (DMSO, 400 MHz) δ (ppm) 12.56 (br, 2H), 8.22 (s, 2H), 8.04 (d, J = 8.4 Hz, 4H), 7.87 (d, J = 8.4 Hz, 4H), 7.79 (d, J = 4.8 Hz, 2H), 7.46 (d, J = 4.8 Hz, 2H), 7.32 (t, J = 8.0 Hz, 2H), 7.14 (t, J = 7.2 Hz, 2H), 3.92 (t, J = 6.0 Hz, 4H), 3.56 (t, J = 6.0 Hz, 4H), 3.41–3.36 (m, 16H), 3.18 (s, 6H). 13C NMR (DMSO, 100 MHz) δ (ppm) 167.73, 162.28, 148.28, 137.36, 133.52, 130.16, 130.03, 129.37, 128.69, 127.02, 126.56, 122.35, 122.07, 109.62, 71.74, 71.70, 70.30, 70.09, 68.43, 68.37, 58.50, 42.01; HRMS-ESI: m/z calcd (%) for C48H51N8O8S2: 931.3271 [M + H]+; found: 931.3242; element analysis for C48H50N8O8S2 (%): C 61.88, H 5.37, N 12.14, calculated C 61.92, H 5.41, N 12.03 (Fig. S5–S7†).
Determination of the limits of detection (LOD)
The LOD was calculated based on the fluorescence titration. Sensor 1 was employed at 1 × 10−5 M. The LOD was calculated using the formula 3σ/k, where σ was the standard deviation of blank (10 samples) and k was the slope between intensity difference versus sample concentration (Fig. S13†).Computational details
The ground-state geometry optimization of 1 and its receptor-additive form (1-2F−) were performed on density functional theory (DFT) with the B3LYP/6-31G(d) level of the Gaussian 03 program.42The UV-vis absorptions of compound 1 and its deprotonated species were calculated with the B3LYP/6-31G(d) level of the Gaussian 03 program based on the optimized ground-state geometry.
Results and discussion
Synthesis and photophysical properties of 1
Compound 1 was synthesized through condensation of compound 2 and 2-hydrazinobenzothiazole in refluxing ethanol (Scheme 1). 1H NMR, 13C NMR, 1H–1H COSY, 1H–13C HSQC and high resolution mass spectra confirmed the identity of compound 1 (Fig. S5–S10†). | ||
| Scheme 1 Synthetic routes of sensor 1. | ||
The normalized UV-vis absorption and photoluminescence (PL) spectra of sensor 1 in DMSO were depicted in Fig. 1. Sensor 1 showed an excitation peak at 502 nm and an emission band centered at 582 nm, respectively. The Stokes shift of sensor 1 was 80 nm. The value of fluorescence quantum yields (Φ) of 1 was 0.715 which was measured in DMSO with the dilute solution method using rhodamine B as the ref. 43.
 | ||
| Fig. 1 Absorption (left) and emission (right) spectra of sensor 1 (1 × 10−5 M) in DMSO. | ||
Sensing of fluoride anion
A hydrazone (–CHN–NH–) contained hydrogen donor moiety of NH, which had the tendency for binding of anions, and DPP moiety was an efficient fluorophore. Therefore, the combination of the hydrazone and DPP in a single molecular framework was very useful to create a sensor in which the anion binding properties of a hydrazone could be monitored by following the changes in the spectral properties. We investigated the anion sensing ability of compound 1 with various anions such as F−, Cl−, Br−, I−, H2PO4−, HSO4−, AcO−, ClO4−, CN−, OCN−, SCN−, N3−, NO2−, IO4− and NO3− (as tetrabutylammonium salts) in DMSO solution. The visual inspection with naked-eye and UV light upon the addition of various above-mentioned anions was shown in Fig. 2. A discernible color change from orange to dark cyan and fluorescent orange to non-fluorescent cyan of 1 solution occurred only in the presence of fluoride anion. A discernible color change from orange to red and fluorescent orange to non-fluorescent purple of 1 solution occurred in the presence of dihydrogenphosphate. These results indicated that 1 had the potential to act as specific sensor for fluoride anion.
 | ||
| Fig. 2 Color change induced upon addition of excess equivalents various anions (tetrabutylammonium salts) to 1 (1 × 10−5 M) in DMSO solution under daylight (up) and UV lamp (low). | ||
Selective detection of fluoride anion by absorption, fluorescence, NMR and density functional studies
The absorption spectral titration of sensor 1 was carried out by addition of fluoride anion in DMSO solution. The systematic changes in the absorption spectra of 1 on addition of TBAF in DMSO are shown in Fig. 3a. Upon addition of fluoride anion, the maximum absorption was red-shifted from 502 to 658 nm (Δλ = 156 nm). Two clear isosbestic points at 460 and 538 nm were produced which indicated the binding of fluoride anion to 1. Correspondingly, the color of the solution changed from orange to dark cyan. Both the new absorption band and the color change almost appeared instantaneously after the addition of fluoride ion. To investigate the selective binding of fluoride to 1, absorption spectra of 1 in the presence of various above-mentioned tetrabutylammonium anions were recorded (Fig. 3b). As shown in Fig. 3b, only fluoride ion induced a prominent bathochromic shift of 1, H2PO4− caused only a slight enhancement of the intensity (εmax) of 1, and no obvious changes were observed upon addition of other anions, which clearly indicated that 1 could act as a highly selective sensor for fluoride ion over other anions.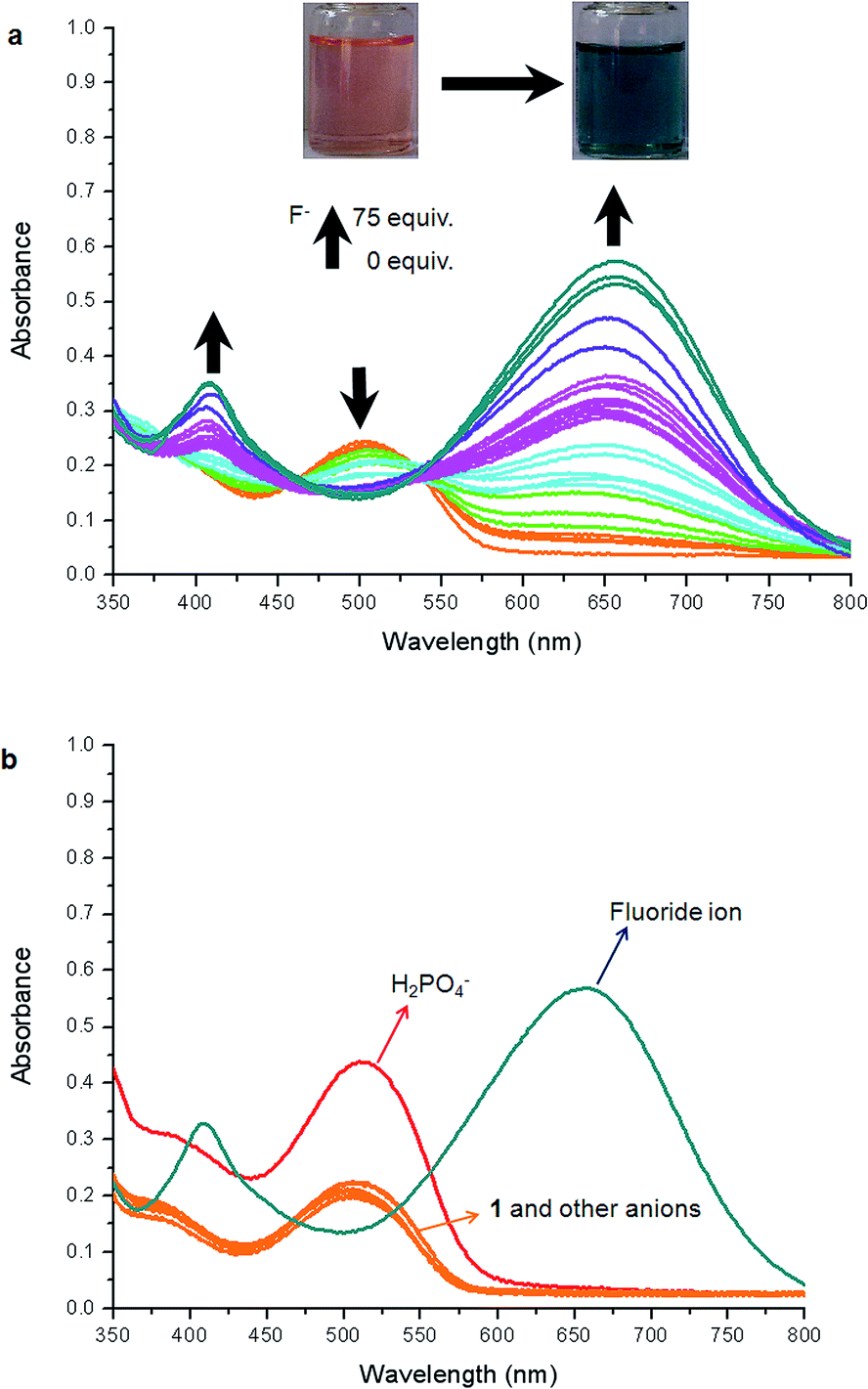 | ||
| Fig. 3 (a) The absorption spectra of 1 (1 × 10−5 M) in DMSO with the addition of different equivalents of fluoride anion. (b) The absorption spectra of 1 (1 × 10−5 M) addition of 75 equiv. of various anions (as TBA + salts) in dry DMSO solution. | ||
The selective sensing of 1 toward fluoride anion was also followed systematically by fluorescence titration. The systematic changes in emission spectra of 1 on addition of TBAF in DMSO were shown in Fig. 4. Upon addition of increasing amounts of fluoride anion, the fluorescence intensity of 1 at 582 nm was gradually decreased and an approximately complete fluorescence quenching [quenching efficiency (I0 − I)/I0 × 100% = 99.8%, I0 = fluorescence intensity of sensor, I = fluorescence intensity obtained with fluoride ion] was observed in the presence of 25 equiv. fluoride ion in DMSO (Fig. 4a). To investigate the selective binding of fluoride to 1, fluorescence spectra of 1 in the presence of various above-mentioned tetrabutylammonium anions were recorded (Fig. 4b). As shown in Fig. 4b, only fluoride ion induced a prominent fluorescent intensity change of 1, H2PO4− caused a descent of the spectra, and no obvious changes were observed upon addition of other anions, which clearly indicated that 1 could act as a highly selective sensor for fluoride ion over other anions.
 | ||
| Fig. 4 (a) The emission spectra (λex = 505 nm) of 1 (1 × 10−5 M) in DMSO with the addition of different equivalents of fluoride ion. (b) Fluorescence spectra (λex = 505 nm) of 1 (1 × 10−5 M) upon addition of 75 equiv. of various anions (as TBA + salts) in dry DMSO solution. | ||
The binding stoichiometry of sensor 1 and F− was further proved by Job's plot according to the continuous variations with a total concentration of [F−] + [1] (1.0 × 10−5 M) (Fig. S11†). The maximum fluorescence emission appeared at the ∼0.33 mole ration of [1]/([F−] + [1]), which indicating the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 interaction between fluoride ion and sensor 1.
:1 interaction between fluoride ion and sensor 1.
The binding constants were calculated from the spectral titration data by Benesi–Hildebrand equation:
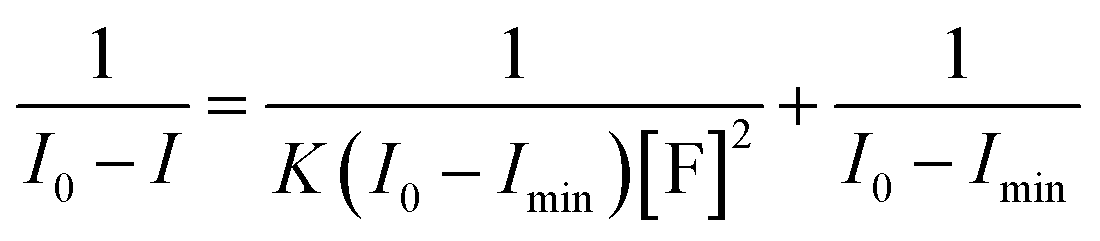 :2 binding mode between sensor and fluoride ion. Linear fitting of the titration profiles resulted in a good linearity (correlation coefficient was over 0.999) (Fig. S12†), which strongly supported the 1:2 binding stoichiometry of sensor 1, and the binding constant was calculated to be 2.69 × 107 M−2 for 1. The quenching constant was found to be 1.7 × 10−3 M−1 for 1 (Fig. S19†). The limits of detection (LOD) of sensor 1 for fluoride ion was further evaluated by using the linear dynamic response and the LOD was found to be 4.07 × 10−8 M for 1 (Fig. S13†). Moreover, the reactivity of sensor 1 toward fluoride was very rapidly (Fig. S14†). These features indicated that sensor 1 might become excellently sensitive sensors with a real-time colorimetric fluorescent response.
:2 binding mode between sensor and fluoride ion. Linear fitting of the titration profiles resulted in a good linearity (correlation coefficient was over 0.999) (Fig. S12†), which strongly supported the 1:2 binding stoichiometry of sensor 1, and the binding constant was calculated to be 2.69 × 107 M−2 for 1. The quenching constant was found to be 1.7 × 10−3 M−1 for 1 (Fig. S19†). The limits of detection (LOD) of sensor 1 for fluoride ion was further evaluated by using the linear dynamic response and the LOD was found to be 4.07 × 10−8 M for 1 (Fig. S13†). Moreover, the reactivity of sensor 1 toward fluoride was very rapidly (Fig. S14†). These features indicated that sensor 1 might become excellently sensitive sensors with a real-time colorimetric fluorescent response.
Further, to examine whether compound 1 could work under basic condition, compound 1 was treated with F− in different concentration of OH− anions, which was dissociated from tetrabutylammonium hydroxide (TBAOH) (Fig. 5 and S20†). As shown in Fig. 5, as the concentration of OH− increased to 1.0 mM, the absorption spectra kept the same, similar to the situation that we observed in emission (Fig. S20†). The absorption maximum was blue-shifted from 502 to 430 nm when the concentration of TBAOH reached to 2.0 mM, and the color of the solution changed from orange to dark yellow. These results indicated that the detection of F− could not only be conducted under the neutral condition but also under the basic environment. In other words, the sensor 1 could distinguish F− anion from a series of anions, even the hydroxide anion.
 | ||
| Fig. 5 The absorption spectra of sensor 1 (1 × 10−5 M) in DMSO in the presence of different concentrations of OH− followed by the addition of 75 equiv. of fluoride anion in DMSO. | ||
To study the interaction of compound 1 with F− ion, we investigated UV-vis responses in detail (Fig. S15†). Only slight enhancement of the band at 658 nm was observed on addition of 0–2 equiv. of F− (Fig. S15a†), which was due to the hydrogen bonding between F− ion and the hydrazone N–H group.44 With further addition of F−, the absorption at 658 nm began to increase significantly and reached the limit value after 75 equiv. of F− were added, indicating that a fluoride-induced deprotonation of thiourea N–H had occurred.45 This was confirmed by adding OH− (as Bu4NOH) to the solution of 1, which gave similar UV-vis spectral changes (Fig. S16†) to those observed with F− ion. The aforementioned results suggested that the sensor–fluoride interaction was a two-step process: (1) at lower fluoride concentration (0–2 equiv.), the N–H⋯F hydrogen bonding occurred; and (2) with increasing fluoride concentration (11–90 equiv.), excess fluoride interacted with the sensor–fluoride complex and led to the deprotonation of the sensor.44 Moreover, competitive ion study of F− ion in presence of all of other anions was also examined (Fig. 6 and S21†). For compound 1, in the presence of 75 equiv. of foreign competing anions, only slight enhancement of the band at 658 nm was observed on addition of 0–200 equiv. of F−. That was, the sensitivity of F− ion was repressed remarkably when 200 equiv. of F− was added to the solution of 1 in the presence of 75 equiv. of the foreign competing anions. And more F− ions (300 equiv.) were needed to deprotonate 1 completely (Fig. 6). In a further study, 75 equiv. of each competing anions were added to the deprotonated system. The results showed that the anions HSO4− could inhibit the deprotonation greatly, while other anions did not induce changes (Fig. S21†). Hence, anions HSO4− should be the repressive factors for the deprotonation process by providing protons to the deprotonated receptor.
 | ||
| Fig. 6 UV-vis absorption spectra of 1 (1 × 10−5 M) after addition of 75 equiv. of representative anions (Cl−, Br−, I−, H2PO4−, HSO4−, AcO−, ClO4−, CN−, OCN−, SCN−, N3−, NO2−, IO4− and NO3−) and then 1−300 equiv. of F− in 75 equiv. aliquots. | ||
The fluoride ion binding to sensor 1 was also monitored by 1H NMR titration studies in DMSO–d6. The systematic changes in 1H NMR signals of 1 upon addition of increasing amounts of fluoride ion were shown in Fig. 7 and S17.† The signal of proton on the hydrazone N–H (Hh) at 12.53 ppm was disappeared significantly upon addition of 0.25 equiv. of fluoride (Fig. S17†). Upfield shifts were observed for all the aromatic and pyridyl protons with the increase of the concentration of fluoride. Furthermore, a new triplet resonance at 16.13 ppm with a coupling constant J = 120 Hz appeared, which was due to the unbound bifluoride ion [HF2]− when 1.0 equiv. of fluoride was introduced. Control experiment showed that the typical signal of [HF2]− also appeared in the DMSO–d6 solution of TBAF (Fig. S17g†) without the sensor, indicating that the [HF2]− ion could also be generated through deprotonation of the solvent by TBAF itself.46 The deprotonation of the hydrazone N–H group was supported by the fact that CHN and aromatic protons showed distinct upfield (Ha, 0.22; Hb, 0.09; Hc, 0.31; Hd, 0.44; He, 0.46; Hf, 0.35; Hg, 0.41 ppm) shifts when 2.0 equiv. of fluoride was introduced compared with the free sensor, arising from an overall change of the electron distribution in the chromophore when the hydrazone N–H group was deprotonated. The disappearance of the Hh signal was owing to the hydrogen bonding between this proton and excess F− (Fig. 7 and S17†). The deprotonation process was completed within addition of 2.0 equiv. of fluoride as no further changes were observed with the addition up to 5.0 equiv. of fluoride.
 | ||
| Fig. 7 Partial 1H NMR titration spectra of sensor 1 (4.7 × 10−3 M) upon addition of increasing amounts of fluoride (TBAF) ion (0–5 equiv.) in DMSO–d6. | ||
Furthermore, 19F NMR titration spectra also provided clear evidence for interaction of F− ion with sensor 1 upon titration with increasing equivalents of F− ion in DMSO–d6 as shown in Fig. S18.† Upon addition of 1.0 equiv. of fluoride to sensor 1, the sharp signal of the free fluoride at −106.7 ppm was downfield to −96.8 ppm, a typical result of hydrogen bonding of fluoride.47 This significant downfield shift 9.9 ppm in 19F NMR spectrum clearly supported the deprotonation of the hydrazone N–H group upon interaction of sensor 1 with F− ion, which was in agreement with the 1H NMR titration experiment. However, the signal of the expected hydrogen bound fluoride did not appear which might be a result of fast proton exchanging between the bound fluorides and the free ones.48
In order to obtain insight into the anion–analyte interaction mechanism, and in particular, to determine whether the recognition process in the system was mediated through the hydrogen-bonding interaction between sensor 1 and F− or proton abstraction from hydrazone N–H of sensor 1 to the fluoride ion, the theoretical calculations had been performed exploiting the density function theory (DFT) calculations.42 Experimental results showed that 2 equiv. of fluoride ion bind with 1 equiv. of sensor, and so the optimization had been performed with the 1:2 receptor-additive forms (sensor-2F−). The structures of sensor and sensor-2F− were optimized at the B3LYP/6-31G(d) level of theory (Fig. S22†). Calculation results showed that the H–F distance was 0.98 Å and the N–H distance was 1.62 Å. And normal H–F bond and N–H bond distances were generally around 0.92 Å and 0.98 Å, respectively, whereas a typical H⋯F hydrogen-bond distance ranges from 1.73 to 1.77 Å. So, one could conclude from these data the deprotonation of the acidic hydrazone N–H protons of sensor 1 and the formation of HF. Additionally, for sensor 1, the phenyl ring was tilted by about 29° toward the DPP core, and the benzothiazolehydrazone moiety was coplanar with the phenyl moiety. For deprotonated species, the phenyl ring was tilted by about 25° toward the DPP core, and the benzothiazolehydrazone moiety was coplanar with the phenyl moiety.
The bathochromic shift of maximum absorption of sensor 1 when binding with fluoride could further be understood in terms of decrease of the potential energy of its HOMO and LUMO. The HOMO and LUMO orbitals of sensor 1 and deprotonated species were shown in Fig. 8. For sensor 1, the HOMO was distributed on the DPP core, hydrazone moiety and benzothiazole moiety, while the LUMO was more distributed on the DPP core and hydrazone moiety. For the deprotonated species, the HOMO was mostly located on both the hydrazone moiety and benzothiazole moiety, while the LUMO was more distributed on both the DPP core and the hydrazone moiety. As described above that the LUMO was the electron deficient in the case of sensor 1, and fluoride would prefer it more rather than the HOMO. Hence, on binding with fluoride the potential energy of the LUMO was raised to comparatively lesser extent than that of the HOMO, which was less preferred by fluoride. This led to narrowing of the energy gap between HOMO and LUMO, which was ultimately responsible for the bathochromic shift of maximum absorption of sensor 1 on binding with fluoride. The theoretically predicted absorption bands for sensor 1 and its deprotonated species were at 567 and 750 nm, respectively; while the experimental observations were at 504 and 582 nm, respectively. The discrepancy between the calculated absorption wavelength and the experimental results was in line with the known fact that the DFT calculations usually underestimate the excitation energy for the transitions with remarkable ICT character.49,50
 | ||
| Fig. 8 HOMO–LUMO orbitals of sensor 1 and its deprotonated species, and energy level diagrams of HOMO and LUMO orbitals of sensor 1 and its deprotonated species calculated on the DFT level using a B3LYP/6-31G* level of theory. | ||
Conclusions
In summary, a new colorimetric and on–ff fluorescence sensor 1 with diketopyrrolopyrrole (DPP) fluorophore for fluoride anion was synthesized with simple procedure. Sensor 1 displayed a rapid responsivity and high selectivity and sensitivity towards F− over other anions, and it also exhibited a very low detection limit of 4.07 × 10−8 M. Meanwhile, it could be easily observed that the sensor for F− ion changed from orange to dark cyan by the naked eye, and from fluorescence orange to non-fluorescence cyan under UV lamp immediately after the F− ion was added. The experimental results revealed that the formation of anionic species from deprotonation of the hydrazone moiety by fluoride ion was responsible for the spectral changes. Density function theory calculations were conducted to rationalize the optical response of the sensor.Acknowledgements
This work was supported by NSFC of China (21171069), the Scientific Research Foundation of University of Jinan (XKY1416), and University Young Key Teacher Home Visit by the Ministry of Education of Shandong Province.References
- G. L. Waldbott, Clin. Toxicol., 1981, 18, 531 CrossRef CAS PubMed.
- S. Matuso, K.-I. Kiyomiya and M. Kurebe, Arch. Toxicol., 1998, 72, 798 CrossRef.
- S.-W. Zhang and T. M. Swager, J. Am. Chem. Soc., 2003, 125, 3420 CrossRef CAS PubMed.
- B. L. Riggs, Bone Miner. Res., 1984, 2, 366 CAS.
- M. Laisalmi, H. Kokki, A. Soikkeli, H. Markkanen, A. Yli-Hankala, P. Rosenberg and L. Lindgren, Acta Anaesthesiol. Scand., 2006, 50, 982 CrossRef CAS PubMed.
- C. Zhang and K. S. Suslick, J. Am. Chem. Soc., 2005, 127, 11548 CrossRef CAS PubMed.
- M. E. Moragues, R. Martínez-Máñez and F. Sancenón, Chem. Soc. Rev., 2011, 40, 2593 RSC.
- R. Martínez-Máñez and F. Sancenón, Chem. Rev., 2003, 103, 4419 CrossRef PubMed.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936 RSC.
- E. B. Veale and T. Gunnlaugsson, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2010, 106, 376 RSC.
- Y. Zhou, J. F. Zhang and J. Yoon, Chem. Rev., 2014, 114, 5511 CrossRef CAS PubMed.
- H. Zollinger, Color Chemistry: Syntheses, Properties, and Applications of Organic Dyes and Pigments; Wiley: Weinheim, Germany, 2003 Search PubMed.
- S. Luňák Jr, M. Vala, J. Vyňuchal, I. Ouzzane, P. Horáková, P. Možíšková, Z. Elíáš and M. Weiter, Dyes Pigm., 2011, 91, 269 CrossRef PubMed.
- B. Wang, N. He, B. Li, S. Jiang, Y. Qu, S. Qu and J. Hua, Aust. J. Chem., 2012, 65, 387 CAS.
- Y. Zhu, A. R. Rabindranath, T. Beyerlein and B. Tieke, Macromolecules, 2007, 40, 6981 CrossRef CAS.
- D. Cao, Q. Liu, W. Zeng, S. Han, J. Peng and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2395 CrossRef CAS.
- A. R. Rabindranath, Y. Zhu, I. Heim and B. Tieke, Macromolecules, 2006, 39, 8250 CrossRef CAS.
- Y. Zhu, I. Heim and B. Tieke, Macromol. Chem. Phys., 2006, 207, 2206 CrossRef CAS.
- X. Zhang, L. J. Richter, D. M. DeLongchamp, R. J. Kline, M. R. Hammond, I. McCulloch, M. Heeney, R. S. Ashraf, J. N. Smith, T. D. Anthopoulos, B. Schroeder, Y. H. Geerts, D. A. Fischer and M. F. Toney, J. Am. Chem. Soc., 2011, 133, 15073 CrossRef CAS PubMed.
- L. Deng, W. Wu, H. Guo, J. Zhao, S. Ji, X. Zhang, X. Yuan and C. Zhang, J. Org. Chem., 2011, 76, 9294 CrossRef CAS PubMed.
- G. Zhang, H. Li, S. Bi, L. Song, Y. Lu, L. Zhang, J. Yu and L. Wang, Analyst, 2013, 138, 6163 RSC.
- G. Zhang, L. Wang, X. Cai, L. Zhang, J. Yu and A. Wang, Dyes Pigm., 2013, 99, 779 CrossRef CAS PubMed.
- Y.-H. Jeong, C.-H. Lee and W.-D. Jang, Chem.–Asian J., 2012, 7, 1562 CrossRef PubMed.
- T. Yamagata, J. Kuwabara and T. Kanbara, Tetrahedron Lett., 2010, 51, 1596 CrossRef CAS PubMed.
- S. Lin, S. Liu, H. Zou, W. Zeng, L. Wang, R. Beuerman and D. Cao, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3882 CrossRef CAS.
- S. Schutting, S. M. Borisov and I. Klimant, Anal. Chem., 2013, 85, 3271 CrossRef CAS PubMed.
- Y. Qu, J. Hua and H. Tian, Org. Lett., 2010, 12, 3320 CrossRef CAS PubMed.
- Y. Qu, S. Qu, L. Yang, J. Hua and D. Qu, Sens. Actuators, B, 2012, 173, 225 CrossRef CAS PubMed.
- G. Zhang, L. Wang, X. Cai, L. Zhang, J. Yu and A. Wang, Dyes Pigm., 2013, 98, 232 CrossRef CAS PubMed.
- M. V. R. Raju and H.-C. Lin, Org. Lett., 2013, 15, 1274 CrossRef CAS PubMed.
- C. Yang, M. Zheng, Y. Li, B. Zhang, J. Li, L. Bu, W. Liu, M. Sun, H. Zhang, Y. Tao, S. Xue and W. Yang, J. Mater. Chem. A, 2013, 1, 5172 CAS.
- H. Guo, W. Liu, R. Sheng, F. Wang, P. Wang and S. Wu, Imaging Sci. Photochem., 2008, 26, 468 CAS.
- X. Zhuang, W. Liu, J. Wu, H. Zhang and P. Wang, Spectrochim. Acta, Part A, 2011, 79, 1352 CrossRef CAS PubMed.
- K. K. Upadhyay, R. K. Mishra, V. Kumar and P. K. R. Chowdhury, Talanta, 2010, 82, 312 CrossRef CAS PubMed.
- S. S. Razi, R. Ali, P. Srivastava, M. Shahid and A. Misra, RSC Adv., 2014, 4, 22308 RSC.
- M. Shahid, P. Srivastava and A. Misra, New J. Chem., 2011, 35, 1690 RSC.
- W. Huang, H. Lin, Z. Cai and H. Lin, Talanta, 2010, 81, 967 CrossRef CAS PubMed.
- Q. Li, Y. Yue, Y. Guo and S. Shao, Sens. Actuators, B, 2012, 173, 797 CrossRef CAS PubMed.
- X. Yang, L. Zheng, L. Xie, Z. Liu, Y. Li, R. Ning, G. Zhang, X. Gong, B. Gao, C. Liu, Y. Cui, G. Sun and G. Zhang, Sens. Actuators, B, 2015, 207, 9 CrossRef CAS PubMed.
- X. Yang, L. Xie, R. Ning, X. Gong, Z. Liu, Y. Li, L. Zheng, G. Zhang, B. Gao, Y. Cui, G. Sun and G. Zhang, Sens. Actuators, B, 2015, 210, 784 CrossRef CAS PubMed.
- Y. Jin, Y. Xu, Y. Liu, L. Wang, H. Jiang, X. Li and D. Cao, Dyes Pigm., 2011, 90, 311 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakr-zewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision C02, Gaussian Inc.; Pittsburgh, PA, 2004 Search PubMed.
- X.-F. Zhang, Y. Zhang and L. Liu, J. Lumin., 2014, 145, 448 CrossRef CAS PubMed.
- D. E. Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, Org. Biomol. Chem., 2005, 3, 1495 Search PubMed.
- S. Kumar, V. Luxami and A. Kumar, Org. Lett., 2008, 10, 5549 CrossRef CAS PubMed.
- C. Jia, B. Wu, J. Liang, X. Huang and X.-J. Yang, J. Fluoresc., 2010, 20, 291 CrossRef CAS PubMed.
- K. M. K. Swamy, Y. J. Lee, H. N. Lee, J. Chun, Y. Kim, S.-J. Kim and J. Yoon, J. Org. Chem., 2006, 71, 8626 CrossRef CAS PubMed.
- G. Xu and M. A. Tarr, Chem. Commun., 2004, 1050 RSC.
- C. Wu, S. Tretiak and V. Y. Chernyak, Chem. Phys. Lett., 2007, 433, 305 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, J. Am. Chem. Soc., 2004, 126, 4007 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra14639e |
| This journal is © The Royal Society of Chemistry 2015 |