
Transcriptome analysis reveals the oxidative stress response in Saccharomyces cerevisiae†
Abstract
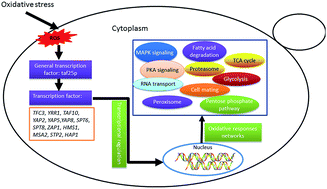
The oxidation stress tolerance of Saccharomyces cerevisiae was improved by global transcription machinery engineering in our previous work. To explore the global perturbation induced by the mutant transcription factor Taf25, an RNA-Seq based gene expression analysis was conducted. Compared to the control strain, a total of 1006 genes with significantly differential expression levels were identified in the mutant strain taf25-3 upon oxidation stress. Fifteen transcription factor-encoding genes were determined, most of which displayed consistent up-regulated signature expressions in response to the challenge of 2 mM H2O2. Based on GO and KEGG enrichment analysis, the identified genes were involved in many metabolic pathways including carbon metabolism, fatty acid degradation, peroxisomal, and synthesis of several amino acids. The genes related to MAP kinase and cAMP-dependent protein kinase A (PKA) signaling pathways were also enriched significantly. The results suggested that the MAP kinase and PKA signaling pathways could be involved in mediation of yeast tolerance against oxidation stress, especially the mating regulation module in MAP kinase which could be reprogrammed by mutation of transcription factor Taf25 in yeast.
 Please wait while we load your content...
Please wait while we load your content...

