
Bacteria microarrays as sensitive tools for exploring pathogen surface epitopes and recognition by host receptors
Abstract
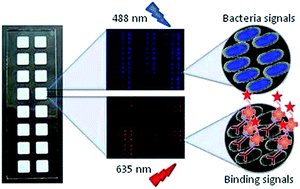
We describe a protocol for the generation and validation of bacteria microarrays and their application to the study of specific features of the pathogen's surface and interactions with host receptors. Bacteria were directly printed on nitrocellulose-coated glass slides, using either manual or robotic arrayers, and printing quality, immobilization efficiency and stability of the arrays were rigorously controlled by incorporating a fluorescent dye into the bacteria. A panel of wild type and mutant strains of the human pathogen Klebsiella pneumoniae, responsible for nosocomial and community-acquired infections, was selected as model bacteria, and SYTO-13 was used as dye. Fluorescence signals of the printed bacteria were found to exhibit a linear concentration-dependence in the range of 1 × 108 to 1 × 109 bacteria per ml. Similar results were obtained with Pseudomonas aeruginosa and Acinetobacter baumannii, two other human pathogens. Successful validation of the quality and applicability of the established microarrays was accomplished by testing the capacity of the bacteria array to detect recognition by anti-Klebsiella antibodies and by the complement subcomponent C1q, which binds K. pneumoniae in an antibody-independent manner. The biotin/AlexaFluor-647-streptavidin system was used for monitoring binding, yielding strain- and dose-dependent signals, distinctive for each protein. Furthermore, the potential of the bacteria microarray for investigating specific features, e.g. glycosylation patterns, of the cell surface was confirmed by examining the binding behaviour of a panel of plant lectins with diverse carbohydrate-binding specificities. This and other possible applications of the newly developed arrays, as e.g. screening/evaluation of compounds to identify inhibitors of host–pathogen interactions, make bacteria microarrays a useful and sensitive tool for both basic and applied research in microbiology, biomedicine and biotechnology.
 Please wait while we load your content...
Please wait while we load your content...

