
A solution-processed high-efficiency p-NiO/n-ZnO heterojunction photodetector
Abstract
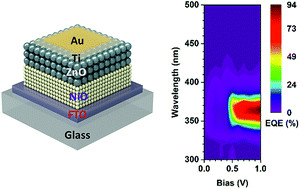
This paper presents a high efficiency heterojunction p-NiO/n-ZnO thin film ultraviolet (UV) photodetector (PD) fabricated on conductive glass substrates. The devices are fabricated by using a simple spin-coating layer-by-layer method from precursor solutions. Photodiodes show good photoresponse and quantum efficiency under UV illumination. With an applied reverse bias of 1 V, the devices show maximum responsivity and detectivity of 0.28 A W−1 and 6.3 × 1011 Jones, respectively, as well as high gain with external quantum efficiency (EQE) of over 90%. By employing ultrathin Ti/Au as top UV transparent metal contacts, this architecture allows the PDs to be illuminated either through glass or metal side. Laser beam induced current is used to examine the local variation of EQE providing information on the photoresponse behavior within the device. Optical properties of NiO and ZnO deposits have also been explored.
 Please wait while we load your content...
Please wait while we load your content...

