
Experimental and theoretical evaluation on the conformational behavior of L-aspartic acid dimethyl ester and its N-acetylated derivative†
Carolyne B. Bragaa,
Lucas C. Ducatib and
Roberto Rittner*a
aPhysical Organic Chemistry Laboratory, Chemistry Institute, University of Campinas, P.O. Box 6154, 13083-970, Campinas, SP, Brazil. E-mail: rittner@iqm.unicamp.br
bChemistry Institute, University of Sao Paulo, 05508-900, São Paulo, SP, Brazil
First published on 4th February 2015
Abstract
In this work the conformational preferences of L-aspartic acid dimethyl ester (AspOMe) and its N-acetylated derivative (AcAspOMe) were evaluated through spectroscopic data and theoretical calculations. Unlike amino acids, their corresponding amino ester derivatives do not exhibit a zwitterionic structure and are soluble in most organic solvents, enabling their studies in these media. Thus, the conformers of AspOMe and AcAspOMe were theoretically determined both in isolated phase and in solution (IEF-PCM model) at the ωB97X-D/aug-cc-pVTZ level. A joint analysis of the experimental and theoretical 3JHH coupling constants in several aprotic solvents allowed assigning the most stable conformers, showing excellent agreement between these approaches. Also, IR spectroscopy allowed us to obtain quantitative data on AcAspOMe conformer populations in different solvents. Natural bond orbital (NBO) analysis indicated that both steric and hyperconjugative contributions count in determining the relative conformer stabilities of these compounds. Intramolecular hydrogen bonding, characterized by Quantum Theory of Atoms in Molecules (QTAIM) and Non-Covalent Interactions (NCI) methodologies, represents only a secondary factor to drive the stabilities of AspOMe and AcAspOMe conformers.
1. Introduction
Amino acids and their derivatives have been under investigation for decades because of the numerous functions that these compounds can perform. Among these studies, a large number is addressed to investigate their conformational preferences,1–3 because these are directly related to the interactions that occur in biological environments and, consequently, with their functions.4,5 However, although significant advances have occurred in both computational and spectroscopic methods, it is still critical to obtain this type of structural information both in isolated phase and in solution.In one way, the remarkable conformational flexibilities of amino acids and the amount of possible intramolecular interactions that can be established between their different functional groups give rise to a large number of low-energy conformers. Thus, gas phase studies are essential, since they provide a unique opportunity to understand the conformer structures as well as their intrinsic properties (such as intramolecular interactions) without the interference of neighboring molecules or the solvent. Nevertheless, the obvious difficulties in experimental studies of isolated amino acids are caused by their high melting points and associated low vapor pressures. They also have low thermal stability, so they tend to decompose before melting.6
In contrast, exploring the conformational preferences of amino acids in solution is essential for a better understanding of their behavior in biological systems. Indeed, a substantial increase in the number of theoretical studies about solvation of amino acids has been recently published.7,8 Notwithstanding, experimental studies in solution are hindered by the fact that these compounds have low solubility in organic solvents, resulting in an additional barrier for experimental studies, including NMR spectroscopy.
In order to overcome the aforementioned difficulties, an alternative approach proposed by our group is to analyze their esterified and N-acetylated derivatives,9–11 which, unlike amino acids, are soluble in most organic solvents. Moreover, these compounds properly simulate the amino acid residue in a polypeptide chain or protein.
Although several studies have been dealt with the conformational preferences of glycine and alanine, amino acids with longer side chains have been less explored, since they present a more complex behavior, such as a larger number of low energy conformers. Specifically, few studies have been performed with L-aspartic acid and their derivatives,1,7 despite their biological importance. There is no doubt that the conformation of residues of this amino acid contributes significantly to the three dimensional shape of polypeptides and proteins, and can change their structures and functions in an active biological system.
Another aspect that is worth mentioning is the lack of detailed evaluation about the driving effects responsible for the conformational preferences of this kind of compounds, since they are limited to identify the number of conformers and their relative energies. Moreover, it is surprising that only intramolecular hydrogen bonding (IHB) is taken into account, while recent studies indicated that the balance between steric and hyperconjugative effects, and not just IHB, are responsible for the conformer stabilities of the amino acids.12–14
Therefore, this study is aimed to investigate the conformational preferences of L-aspartic acid dimethyl ester (AspOMe) and its N-acetylated derivative (AcAspOMe) in the isolated phase as well as in several aprotic solvents and to evaluate the intramolecular interactions responsible for the stabilities of the most stable conformers. For this purpose, theoretical and experimental 3JHH coupling constants were used in the analysis of their conformational equilibra. Also, infrared data were employed as a complement to determine AcAspOMe populations. Quantum Theory of Atoms in Molecules (QTAIM), Non-Covalent Interactions (NCI) and Natural Bond Orbital (NBO) analysis have also been carried out for the interpretation of the obtained results.
2. Experimental section
2.1. Synthesis of the compounds
AspOMe was commercially available as a hydrochloride and was deprotonated using activated zinc powder.15 The synthesis of AcAspOMe consisted in the esterification of N-acetyl-L-aspartic acid. The detailed procedures are described in the ESI.†2.2. NMR spectra
1H NMR spectra were recorded on a Bruker Avance III spectrometer operating at 600.17 MHz for 1H. Spectra were obtained using solutions of ca. 15 mg in 0.7 mL of deuterated solvents (C6D6, CDCl3, CD2Cl2, acetone-d6, CD3CN and DMSO-d6), referenced to internal TMS. The typical conditions used were: probe temperature of 25 °C, 16 transients, spectral width around 6.0 kHz and 64k data points with an acquisition time of ca. 6 s. The free induction decays (FID) were zero-filled to 128k, providing a digital resolution of 0.09 Hz per point.2.3. Infrared spectra
Infrared spectra were recorded on a Shimadzu FT-IR Prestige-21 spectrometer using samples with a concentration of 0.03 mol L−1 and a NaCl cell with an optical path of 0.5 mm. The solutions were prepared with solvents of different polarities: CCl4, CHCl3, CH2Cl2 and CH3CN. The spectra were acquired with 64 scans and resolution of 1 cm−1. The overlapped carbonyl bands were deconvoluted by means of the GRAMS curve fitting program. The equipment was purged with dry nitrogen gas.2.4. Theoretical calculations
The conformational investigation to find the possible energy minima of AspOMe was performed starting from the six conformers (I, III, IV1, IV2, V1 and V2) previously obtained for alanine methyl ester (AlaOMe),9 which have the less energetic arrangement of the backbone [CH3–O–C(O)–CH(NH2)–]. To this end, a methyl hydrogen atom (side chain) of AlaOMe was replaced by the C(O)–O–CH3 group, giving rise to the side chain of aspartic acid. Thus, six potential energy surfaces (PES, Fig. S1 in the ESI†) were built from these six new geometries by simultaneously scanning the χ1 [C(O)–C–C–C(O)] and χ2 (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–C–C) dihedral angles (Fig. 1a) in 36 steps from 0° to 360°, at B3LYP/cc-pVDZ level. In this stage, the earlier optimized ϕ [nN–N–C–C(O)] and ψ [N–C–CO] dihedral angles were kept fixed to preserve the optimized geometry of the main chain.
C–C–C) dihedral angles (Fig. 1a) in 36 steps from 0° to 360°, at B3LYP/cc-pVDZ level. In this stage, the earlier optimized ϕ [nN–N–C–C(O)] and ψ [N–C–CO] dihedral angles were kept fixed to preserve the optimized geometry of the main chain.
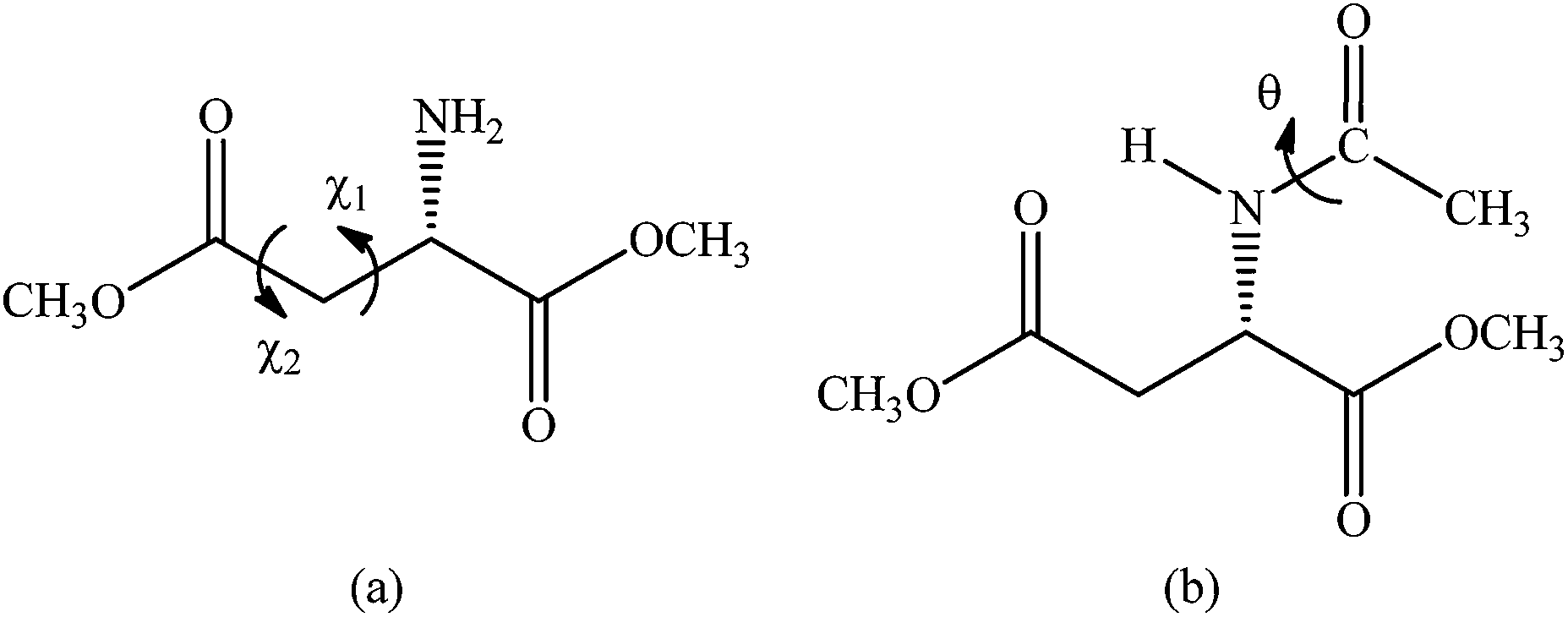 | ||
| Fig. 1 Analyzed dihedral angles: (a) χ1 [C(O)–C–C–C(O)] and χ2 (OC–C–C) for AspOMe and (b) θ [C–C(O)–N–C] for AcAspOMe. | ||
The 34 minima resulting from the PES were subsequently fully reoptimized using DFT different functionals (B3LYP,16 CAM-B3LYP,17 M05-2X,18 M06-2X,19 B97-D20 and ωB97X-D21) with the aug-cc-pVTZ basis set, and their harmonic frequencies were calculated and also zero-point energy (ZPE) correction. The obtained data were referenced to the single point calculations at the MP2/aug-cc-pVTZ level of theory. Some conformers were discarded, since they: (i) have imaginary harmonic frequencies (are not true energy minima) or (ii) do not present significant contribution to the conformational equilibrium of the compound in isolated and including the solvent effect, resulting in 8 stable conformers for AspOMe.
After, these lowest energy geometries found for the amino ester were used as starting points to determine the AcAspOMe conformers. For each previously optimized AspOMe geometry, the N-acetyl group was added by replacing one of the hydrogen atoms of the amine group, giving rise to an amide linkage, and thus eight potential energy curves (PEC) were obtained by rotating the θ [C–C(O)–N–C] dihedral angle (Fig. 1b), at the B3LYP/cc-pVDZ level. Each PEC presented two stereoisomers (cis and trans); hence the sixteen minima geometries found were fully reoptimized at the ωB97X-D/aug-cc-pVTZ level, which showed, for AspOMe, appreciable correlation with the MP2 one, and their frequencies were calculated with ZPE correction.
Thereafter, the resulting conformers of both compounds were fully optimized by using the IEF-PCM (Integral Equation Formalism Polarizable Continuum Model)22 in aprotic solvents of different dielectric constants, at the ωB97X-D/aug-cc-pVTZ level. Also, from these IEF-PCM calculations the 3JHH coupling constants were obtained for each conformer using the ωB97X-D functional and the EPR-III23 basis set for the hydrogen and carbon atoms, whereas oxygen and nitrogen were represented by aug-cc-pVTZ basis set, since this level reproduces 3JHH with good accuracy.24
Lastly, natural bond orbital (NBO)25 analysis was performed on the same ωB97X-D/aug-cc-pVTZ level, as well as QTAIM and NCI calculations. All calculations employed the Gaussian09 program package, Revision D.01,26 excepting the QTAIM and NCI, which were carried out with AIMALL27 and NCIPLOT 3.0 (ref. 28) programs, respectively.
3. Results and discussion
3.1. AspOMe
The eight most stable geometries found in the PES (Fig. S1 in the ESI†) were optimized using different DFT methods and ab initio MP2 with aug-cc-pVTZ basis set. A complete comparison between the energies, relative energies and investigated dihedral angles obtained using these different methods is shown in Table S1 (ESI†). It was observed that all methods gave values of the dihedral angles very close to the ones from the MP2 method, which was used as the reference. However, the energies varied considerably with the theoretical level used. On the one hand, the popular B3LYP, as well as CAM-B3LYP, showed the most discrepant values related to the MP2, but a good correlation was obtained for the ωB97X-D functional compared with the reference, which has also been the method used in a previous work of the corresponding derivatives of the amino acid L-proline.10 Therefore, ωB97X-D, which includes empirical atom–atom dispersion corrections important to describe non-covalent interactions (such as hydrogen bonding), was used as the main computational method for the subsequent calculations.The geometries of the eight most stable conformers involved in the conformational equilibrium of the AspOMe (at ωB97X-D/aug-cc-pVTZ level of theory) are shown in Fig. 2. Each conformer was named with a roman numeral followed by the letters a, b or c. The numbers indicate the order of stability in isolated phase (considering the relative energies with ZPE at ωB97X-D) and the letters represent the relationship between side and main chains, which are depicted on the Newman projections of Fig. 3.
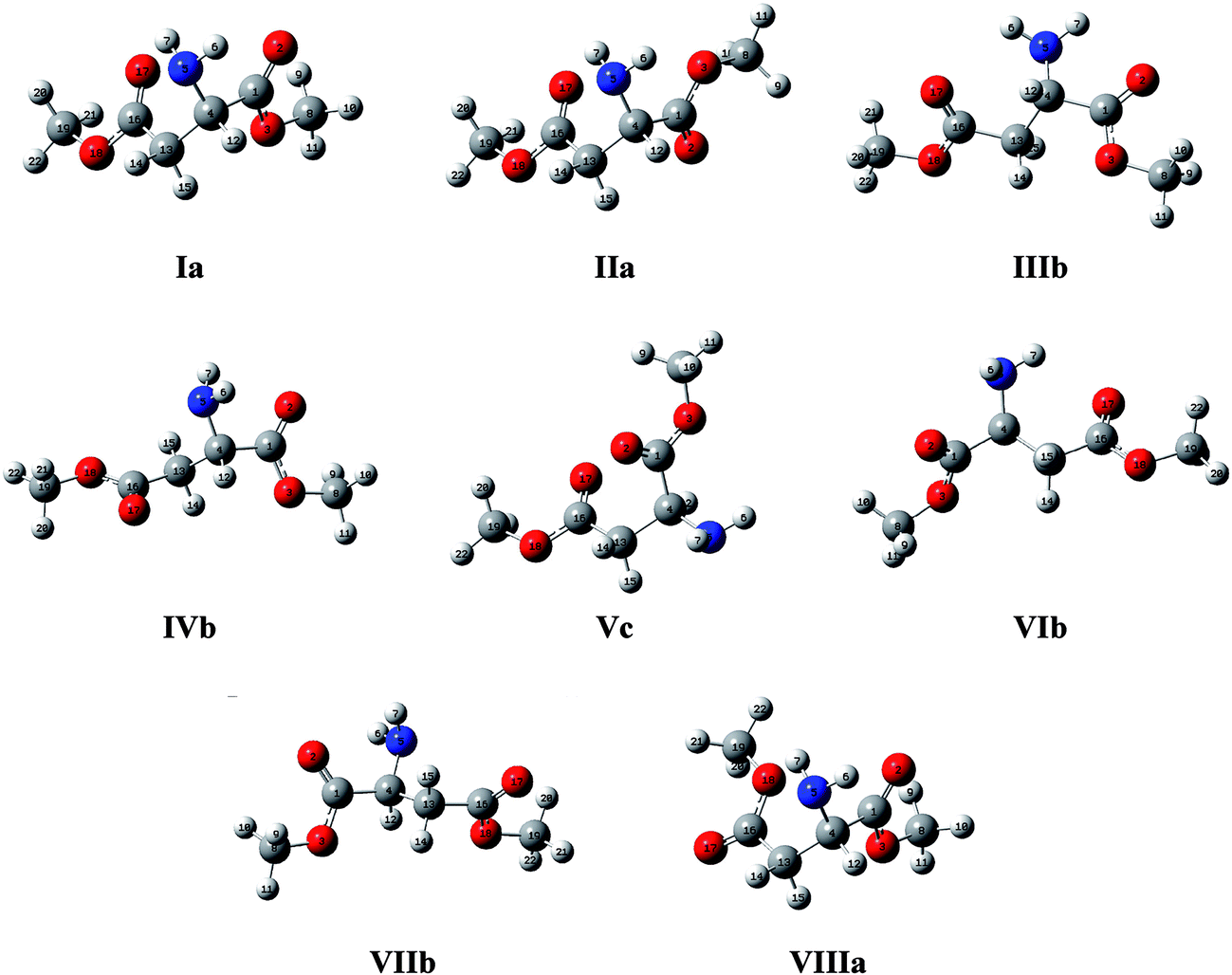 | ||
| Fig. 2 Spacial representation of the most stable conformers of AspOMe, defined by the dihedral angles χ1 and χ2 and obtained at the ωB97X-D/aug-cc-pVTZ level of theory. The conformers are ordered according to their relative energies in isolated phase. | ||
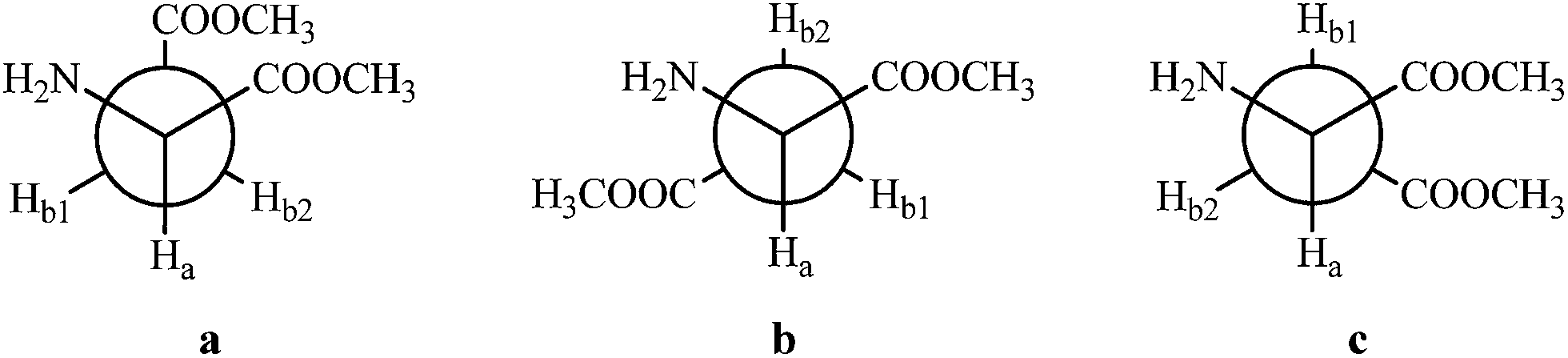 | ||
| Fig. 3 Newman projections showing the dispositions a, b and c of the side chain, resulting from rotation around the Cα–Cβ bond. | ||
The relatively large number of conformers is due to the presence of a polar side chain (R = –CH2COOCH3), which can act as a proton acceptor, multiplying the possible combinations of intramolecular interactions that can be established between the different functional groups. Of these, the conformer Ia is the most stable for the isolated compound and accounts for 30.4% of the conformational population. However, the conformer IIa has also a relatively large stability (26.8%) compared to the remaining conformers, since together with Ia represents more than half of the conformational equilibrium of AspOMe in isolated phase. The others conformers have similar energies and they contribute much less to the overall conformational population.
Inspection of conformers Ia and IIa shows a great similarity in their spatial arrangements, with values of about 60° and 0° for the dihedrals χ1 and χ2, respectively. The only structural difference between these two most stable conformers is the dihedral ψ [N–C–CO], which is syn for the former and anti for the latter. Assuming that these conformers are almost isoenergetics, it can be concluded, therefore, that this dihedral does not affect their stabilities. Then, which effects are related with the observed conformational preferences?
When the conformational equilibra of amino acids and their derivatives are described in literature, rares are the examples that do not explain the observed conformational stabilities in terms of intramolecular hydrogen bonding (IHB). However, recent works published by our research group9,10 have shown that, in many cases, other interactions are responsible for the conformational preferences, as hyperconjugative and steric effects. Therefore, to identify the intramolecular effects that occur in this isolated system, NBO calculations and QTAIM and NCI topological analyses were performed.
The molecular graphs obtained by the QTAIM (Fig. 4) characterize an IHB in conformers Ia, IIa, VIb and VIIIa, which show a bond critical point (BCP) and a bond path (BP). According to Bader, the presence of BCP and BP are necessary and sufficient conditions for two atoms to be considered chemically bonded.29 In addition, the topological criteria30 were also evaluated (Table 1), confirming that the IHB exhibited by QTAIM are stable. Nevertheless, it can be stated that this interaction has a small contribution to the stability of VIb and VIIIa, given their high relative energies.
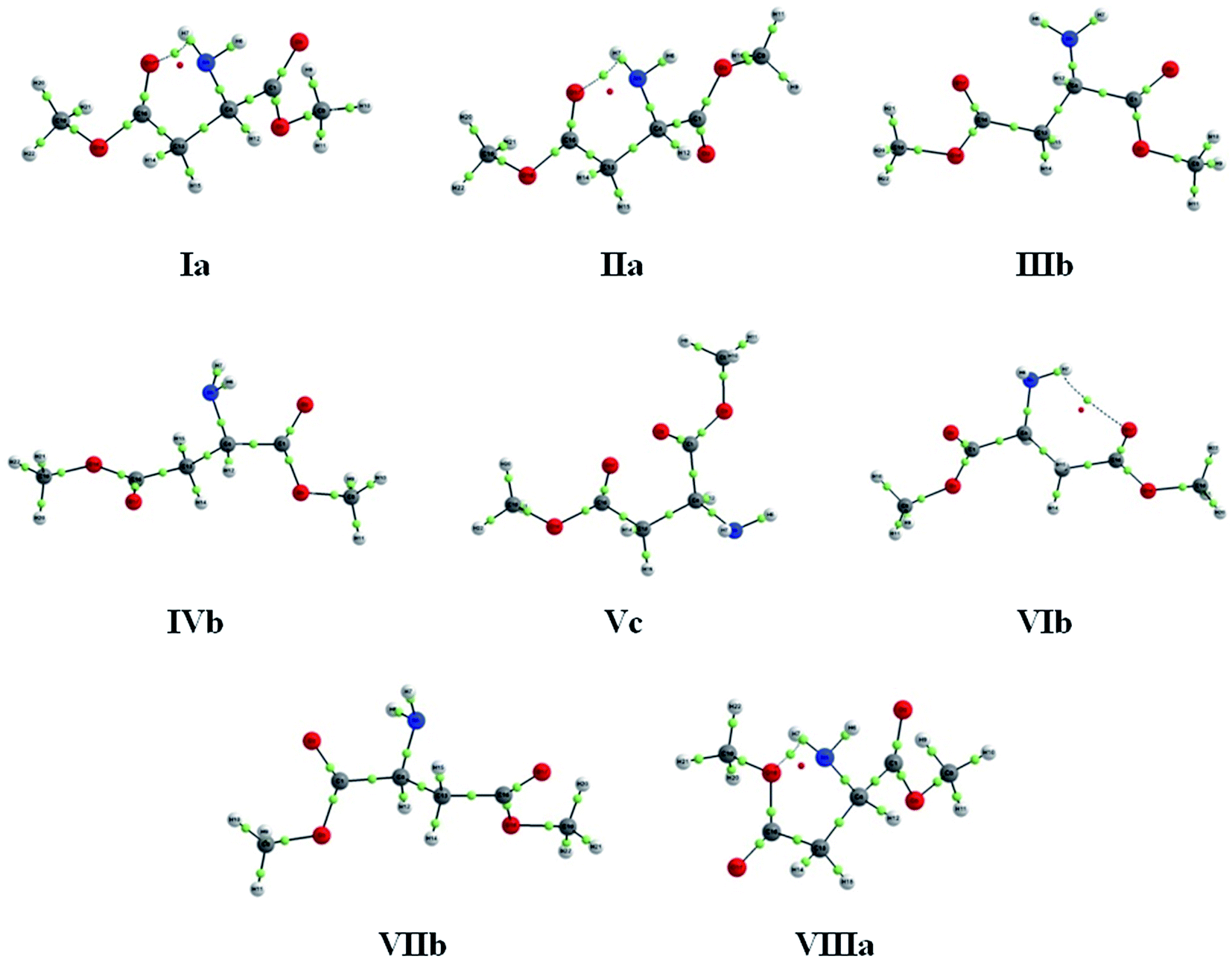 | ||
| Fig. 4 QTAIM molecular graphs for the AspOMe conformers, obtained at the ωB97X-D/aug-cc-pVTZ level of theory. Dotted lines represent the bond paths (BPs), the green dots represent the bond critical points (BCPs) and the ring critical points (RCPs) are indicated by red dots. | ||
| ρHBCPb | ∇2ρ HBCP | q(H7) | E(H7) | M1(H7) | V(H7) | εHBCP | |
|---|---|---|---|---|---|---|---|
| a The numbering of atoms is in Fig. 2; q (Ω) is the atomic charge, E (Ω) is the total energy, M (Ω) is the dipolar polarization and V (Ω) is the atomic volume of given atom.b ρHBCP, ∇2ρHBCP, q(H7), E(H7), M1(H7) and V(H7) in atomic units.c The conformer Vc does not form an IHB and, therefore, the QTAIM properties of H7 of this conformer can be used as reference to evaluate IHB in the others. | |||||||
| Ia | 0.0111 | +0.044 | +0.374 | −0.4804 | 0.173 | 29.02 | 0.6507 |
| IIa | 0.0127 | +0.051 | +0.379 | −0.4784 | 0.172 | 28.32 | 0.3716 |
| Vcc | — | — | +0.355 | −0.4879 | 0.187 | 32.43 | — |
| VIb | 0.0116 | +0.045 | +0.379 | −0.4775 | 0.173 | 28.59 | 0.6575 |
| VIIIa | 0.0114 | +0.047 | +0.366 | −0.4850 | 0.176 | 29.35 | 0.5565 |
Although the QTAIM is a widely used method for characterizing IHB, some studies have shown that it fails in detecting some weak interactions.31,32 Moreover, unlike Bader, other reports in the literature also indicate that BP not necessarily represent a bonding situation.33 Thus, as complement to the results obtained by Bader's theory (QTAIM), the recently developed NCI analysis was also used, which is based on data of electron density gradient and not only in punctual density values, as QTAIM. The plots of the reduced density gradients [s(r)] versus sign (λ2)ρ(r) (Fig. S1 in the ESI†), as well as the gradient isosurfaces (Fig. S2 in the ESI†), were obtained for all conformers of AspOMe. The negative values of sign (λ2)ρ(r) shown in the Fig. S1† are an indication of attractive interactions, in this case the N–H⋯O IHB, which are spotted in blue color on gradient isosurfaces. There are also positive values of sign (λ2)ρ(r), which characterize repulsive interactions and are marked in red color on isosurfaces, and lastly, van der Waals interactions (green) are related to values of sign (λ2)ρ(r) near zero. Therefore, the NCI analysis confirmed the occurrence of IHB in conformers Ia, IIa, VIb and VIIIa and, also, it revealed a weak interaction in IIIb, which was not identified by QTAIM, providing more reliable results.
It is worth mentioning that for the conformations with IHBs, BSSE values could also be considered. However, although significant intramolecular BSSE effects can affect the single molecules, where there can be noncovalent interactions between different parts of the molecule, studies have shown that larger basis sets are used as an approximation to correct intramolecular BSSE.34 Therefore, the basis set applied in this work, the aug-cc-pVTZ, can be considered large enough to include these BSSE effects. Other attempt to correct the intramolecular BSSE is made through using the counterpoise (CP) correction (by dividing the molecule into fragments). The main problem with this simplification of the system is that there is no unique way to define the fragments.35–37 Thus, based on arbitrariness regarding the appropriate correction procedure for the intramolecular BSSE, we find more suitable to use a larger basis set instead using CP correction.
Next, NBO analysis was performed in order to find the contributions of steric and hyperconjugative effects for the energy of each conformer. According to the results of the Table 2, the order of destabilization in relation to the steric effects is: IVb < VIIb < IIIb < VIb < Vc < Ia < IIa < VIIIa. It is evident from these relations that the conformers a are those exhibiting the greatest steric effects, which is consistent, because they have three bulky groups targeted to the same region of space (Fig. 3). Although the conformers a are the most destabilized forms by steric effects, they are also the most stabilized due to hyperconjugations, as follows: VIIb < IVb < IIIb < VIb < Vc < Ia < VIIIa < IIa. In turn, the conformers b are those with the lowest values of Erel,Lewis (the carboxyl groups are on opposite sides, in order to reduce their steric hindrances), but also the lowest Erel,Hyper. Therefore, these results show that an interplay between steric and hyperconjugative effects determines the conformational preferences of this compound, and the conformers Ia and IIa present the most favorable energy balances, contributing to their stabilities in isolated phase.
| Ia | IIa | IIIb | IVb | Vc | VIb | VIIb | VIIIa | |
|---|---|---|---|---|---|---|---|---|
| a Relative energies in kcal mol−1. | ||||||||
| Erel,Tot | 0.00 | 0.15 | 0.63 | 1.10 | 0.93 | 1.37 | 1.50 | 1.42 |
| Erel,Lewis | 5.67 | 6.94 | 0.27 | 0.00 | 4.60 | 3.59 | 0.05 | 7.24 |
| Erel,Hyper | 7.12 | 8.25 | 1.10 | 0.36 | 5.13 | 3.67 | 0.00 | 7.27 |
The energies of the hyperconjugative interactions which most contribute to Erel,Hyper were also analyzed from the NBO calculations (Table S2 in the ESI†). Although there are small energy variations, no major differences are observed among the conformers. So, a specific interaction cannot be pointed as the only responsible for stabilizing a given geometry.
It was also verified the presence of charge transfer interactions between LP2(O17) → σ*N5–H7 in Ia and IIa, as well as LP2(O2) → σ*N5–H7 in IIIb, characteristics of intramolecular hydrogen bonding (IHB). Their energies are 0.50, 0.72 and 0.85 kcal mol−1 in the isolated phase, respectively. However, no hyperconjugation with energy above 0.50 kcal mol−1 was observed in conformers VIb and VIIIa.
Then, the evaluation of the rotational isomerism taking into account the solvent effects was also performed using a joint analysis of NMR spectroscopy and theoretical calculations. It is well known that the vicinal spin–spin coupling constant (3JHH) exhibits a dihedral angle dependence and, therefore, allows us to examine the conformational preferences of different compounds.33 For amino acid residues, specifically, such constant can help in the identification of the most stable conformers by relating the experimental 3JHH with the dihedral angles of the backbone and side chains. However, due to the fast interconversion rate between the conformers, the experimentally observed 3JHH,obs in solution is a weighted average of the contribution of each conformer i (ηix3JHH,i), which can be estimated through the following equation:
| 3JHH,obs = ∑ηix3JHH,i |
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Solvent | εb | δNH | δHa | δHb1 | δHb2 | δHc | δHd | 3JHaHb1 | 3JHaHb2 |
| a Error in the measurements of J = ±0.05 Hz.b Dielectric constant. | |||||||||
| C6D6 | 2.3 | — | 3.89 | 2.88 | 2.76 | 3.25 | 3.28 | 4.2 | 6.0 |
| CDCl3 | 4.8 | 4.54 | 4.26 | 3.21 | 3.12 | 3.73 | 3.80 | 4.2 | 5.6 |
| CD2Cl2 | 9.1 | — | 4.13 | 3.13 | 3.08 | 3.72 | 3.79 | 4.1 | 5.8 |
| Acetone-d6 | 20.7 | — | 4.17 | 3.23 | 3.11 | 3.68 | 3.76 | 4.8 | 5.2 |
| CD3CN | 37.5 | 3.65 | 4.05 | 4.03 | 3.98 | 3.67 | 3.73 | 4.8 | 5.7 |
| DMSO-d6 | 46.7 | — | 3.80 | 3.73 | 2.68 | 3.60 | 3.64 | 6.3 | 6.3 |

The values listed show that the experimental 3JHaHb1 and 3JHaHb2 values vary with the change of solvent indicating, therefore, changes in the conformer populations. In nonpolar solvents, the diastereotopic hydrogens (Hb1 and Hb2) are at very different chemical environments and couple with the alpha-hydrogen (Ha) with different values of 3JHaHb. By increasing the solvent polarity, on the other hand, such constants of Hb1 and Hb2 with Ha exhibit closer values, until in DMSO-d6, the solvent used of higher dielectric constant, they become equal (3JHaHb1 = 3JHaHb2 = 6.3 Hz). Considering that the rotational isomerism of amino acids is usually composed by forms a, b and c, shown in the Newman projections of Fig. 3, the analysis of these three possible side chain arrangements in relation to the main chain reveals that the geometry a shows only gauche dispositions between hydrogens Ha and Hb, while one arrangement anti and one gauche are observed for conformations b and c. For this reason, it is expected that the 3JHaHb1 and 3JHaHb2 values are greater in c and b, respectively, than in a, since anti hydrogens present spin–spin coupling constants larger than gauche hydrogens. Thus, it can be suggested that in solvents with higher dielectric constants there would be a stabilization of arrangements b and c in relation to the forms a, explaining the fact that the experimental vicinal coupling constants have shown larger values.
In order to understand and explain in more detail these assumptions, theoretical calculations with solvent inclusion using the IEF-PCM model were also performed (Table 4) and show that the minimum Ia remains as the most stable, and the increase of dielectric constant of the solvent causes a subtle stabilization in this geometry. At the same time, there is a destabilization of IIa (from 26.8% for the isolated phase to 14.3% in DMSO), which becomes almost isoenergetic with IVb in polar solvents, being the third more stable in solution. Thus, since these three geometries represent about 60% of these conformational equilibria of AspOMe, the experimental 3JHaHb values are mainly due to these three conformers. Small variations in populations are exhibited by the other conformers, verifying that their energies become even closer to each other with the increase of solvent polarities.
| Conformer | μ | Benzene | Chloroform | Dichloromethane | Acetone | Acetonitrile | DMSO | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Erel | P | Erel | P | Erel | P | Erel | P | Erel | P | Erel | P | ||
| a ZPE correction was included. | |||||||||||||
| Ia | 3.19 | 0.00 | 27.6 | 0.00 | 29.3 | 0.00 | 30.7 | 0.00 | 31.4 | 0.00 | 30.5 | 0.00 | 30.5 |
| IIa | 0.41 | 0.16 | 21.1 | 0.28 | 18.2 | 0.40 | 15.8 | 0.43 | 15.3 | 0.43 | 14.7 | 0.45 | 14.3 |
| IIIb | 2.79 | 0.56 | 10.7 | 0.67 | 9.4 | 0.75 | 8.7 | 0.89 | 7.0 | 0.88 | 6.9 | 0.88 | 6.9 |
| IVb | 1.69 | 0.43 | 13.3 | 0.40 | 14.9 | 0.45 | 14.8 | 0.46 | 14.5 | 0.43 | 14.7 | 0.42 | 15.0 |
| Vc | 0.39 | 0.66 | 9.1 | 0.71 | 8.8 | 0.73 | 9.0 | 0.78 | 8.5 | 0.76 | 8.5 | 0.76 | 8.5 |
| VIb | 3.42 | 0.86 | 6.5 | 0.95 | 5.9 | 0.96 | 6.1 | 0.86 | 7.4 | 0.79 | 8.0 | 0.80 | 7.9 |
| VIIb | 3.49 | 0.87 | 6.3 | 0.82 | 7.3 | 0.80 | 8.0 | 0.78 | 8.5 | 0.73 | 8.9 | 0.72 | 9.0 |
| VIIIa | 2.07 | 0.97 | 5.4 | 0.92 | 6.2 | 0.89 | 6.9 | 0.86 | 7.4 | 0.81 | 7.8 | 0.80 | 7.9 |
The 3JHaHb,i coupling constants for the individual conformers were calculated (Fig. 5a), as well as the estimated contributions of each conformer (ηix3JHaHb,i) to 3JHaHb1 and 3JHaHb2 (Fig. 5b) experimentally obtained (see also these values in Table S3 and S4 in the ESI†, respectively). Fig. 5a shows that variations in 3JHaHb,i of each conformer are insignificant, but the results of Fig. 5b support the changes in experimental 3JHaHb. In nonpolar solvents, the experimental 3JHaHb1 is about 4.2 Hz, in agreement with the individual values of this constant for conformers Ia and IIa (approximately 3.3 and 3.7 Hz, respectively), which are among the main responsible for the observed values. In more polar solvents, although these conformers again exhibit the largest contributions to the experimental values, there is a secondary factor that causes the increase of the experimental 3JHaHb. In this case, the increase in the polarity of the medium decreases the difference among the conformer energies, especially those whose forms are b and c. Therefore, it was found that the experimental 3JHaHb values are in agreement with the theoretical results, and the combined use of theoretical calculations and NMR data allowed showing the influence of the solvent on the conformational equilibrium of AspOMe.
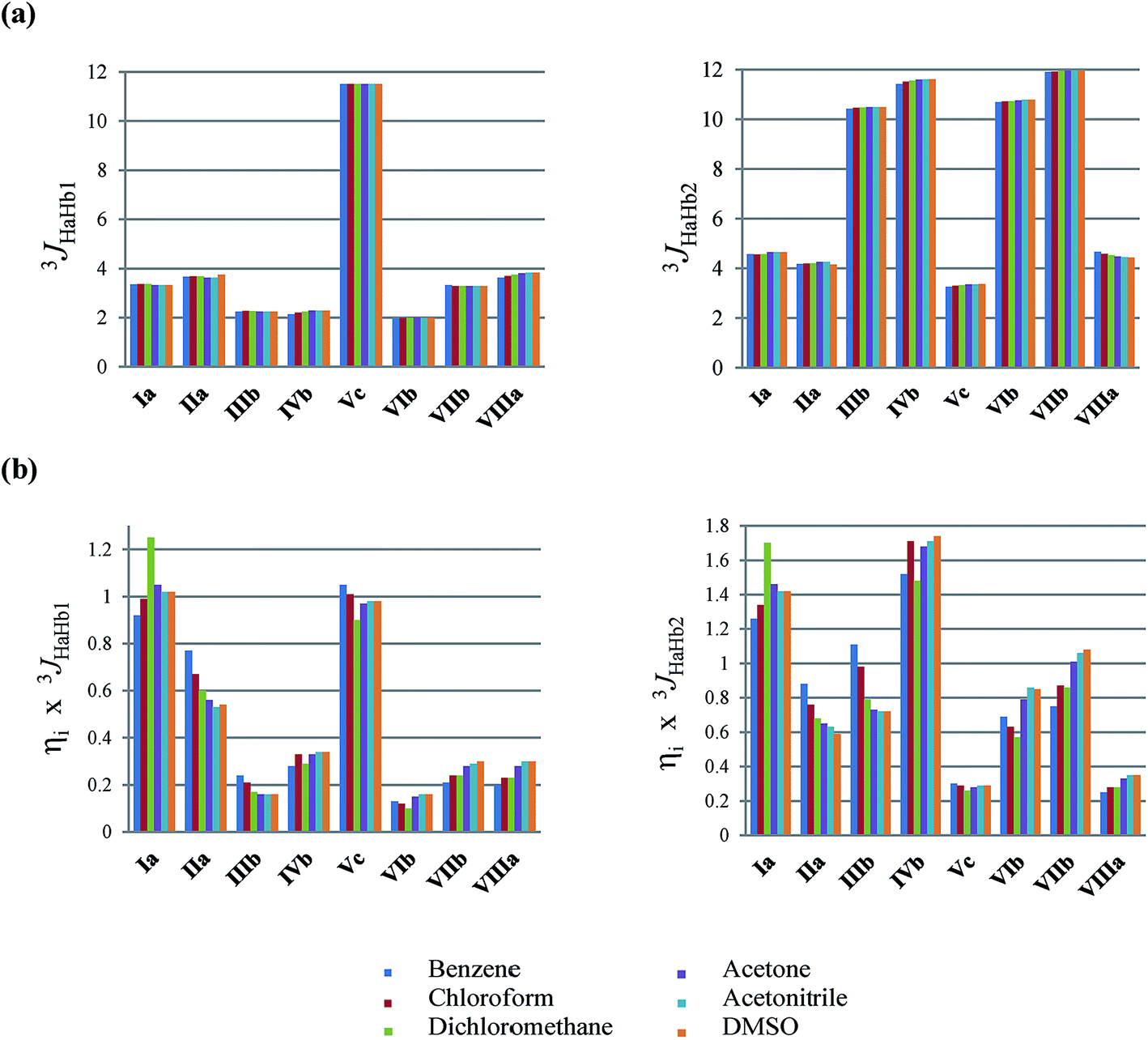 | ||
| Fig. 5 (a) Individual coupling constants 3JHaHb,i for the AspOMe conformers and (b) conformational contributions (ηix3JHaHb,i) for the observed 3JHaHb,obs coupling constants, calculated at the ωB97X-D/EPR-III level of theory, by using the IEF-PCM in several solvents. The values are presented in Hz. | ||
One fact that called our attention was the influence of the solvent on the relative order of stabilities, mainly for IIa and IIIb, since the latter is the third most stable in isolated phase and becomes the most energetic in DMSO. Although IHB and a balance between hyperconjugative and steric effects are the factors governing the conformational preferences of this compound in isolated phase, it is known that intramolecular effects can be disturbed in solution. A well known example is the weakening or even breaking of an IHB due the solvation carried by the solvent. The same is true for the dipole repulsion. So, the conformer interactions in solution were also checked. Table S5 (in the ESI†) shows that the conformer Ia undergoes a small stabilization in more polar solvents, contrary to expectations, which would be increase its energy because weakening (or even the rupture) of the IHB. As much as its energy difference of steric repulsion has increase according the solvent polarity, the same was observed for the stabilizing energy (Erel,Hyper). That is, as well as in isolated phase, the joint contribution of hyperconjugations and steric effects accounts for its greater stability in solution. This is also valid for other conformers, whose relative energies resulting of stabilizing and destabilizing contributions, namely Erel,Tot, exhibit exactly the same trend observed in Table 4.
Thus, based on the data presented for the AspOMe, one can say that both in isolated phase and solution the conformational preferences are governed by a balance between steric and hyperconjugative interactions and, as a side effect, the IHB.
3.2. AcAspOMe
A similar study to that performed for AspOMe was conducted for the AcAspOMe, its N-acetylated derivative. The three more stable conformers of sixteen minima found from PEC are shown in Fig. 6. Their energies, populations and geometric parameters are presented in Table 5. The other conformers will be not mentioned in this discussion, since they have negligible populations (less than 5%) both in isolated phase and solution. | ||
| Fig. 6 Spacial representations of the most stable conformers of AcAspOMe, defined by the θ [C–C(O)–N–C] dihedral angle and obtained at the ωB97X-D/aug-cc-pVTZ level of theory. | ||
| Parameters | trans-Ia | trans-IIIb | trans-Vc |
|---|---|---|---|
| a ZPE correction was included.b Dihedral angles in degrees.c 1 au = 627.5095 kcal mol−1.d The numbering of atoms is in Fig. 6. | |||
| E (hartrees) | −743.42574 | −743.42060 | −743.42052 |
| Erel (kcal mol−1)c | 0.00 | 3.23 | 3.28 |
| P (%) | 100.0 | 0.0 | 0.0 |
| ω [OC1–O–C] |
3.8 | 0.6 | 4.9 |
| ψ [N–C–CO] |
10.3 | 22.3 | 142.9 |
| χ1 [C(O)–C–C–C(O)] | 56.7 | 173.6 | 70.7 |
| χ2 [OC16–C–C] |
17.8 | 55.7 | 32.0 |
| θ [C–C(O)–N–C] |
174.9 | 169.0 | 172.8 |
The name of each conformer was derived from the respective AspOMe starting geometry, including the cis–trans denomination, related to the rotation of θ [C–C(O)–N–C] dihedral angle (Fig. 1b), which indicates the position of the amide methyl group with respect to the C(O)–OCH3. Therefore, the most stable conformers of AcAspOMe are trans, of which the trans-Ia is more stable than the second less energetic (trans-IIIb) by 3.23 kcal mol−1, corresponding to a population of about 100% of the conformational equilibrium, in isolated phase. It is noteworthy that this conformer presents a spatial arrangement very similar to the most stable conformer of AspOMe (Ia, Fig. 2), whose structural difference occurs only in small deviations of the angles due the additional presence of the acetyl group. So, this additional group in the N-acetylated derivative increases the steric repulsion interactions, disadvantaging almost all conformers, except the trans-Ia.
Although steric disturbance in AcAspOMe is significant compared to the AspOMe, other effects, such as IHB and hyperconjugation, are also likely to be important contributions to its observed conformational preferences. In this sense, these interactions were analyzed by QTAIM, NCI and NBO analysis.
The molecular graphs (Fig. 7a) show no BP or BCP referring to an IHB, indicating the absence of this interaction by QTAIM methodology. However, it is observed in trans-Ia isosurface (Fig. 7b), obtained by NCI method, a blue volume between the carbonyl oxygen (side chain) and hydrogen attached to nitrogen, representing an attractive non-covalent interaction, which corresponds to an IHB. The red volume near to this blue volume appears as consequence of the formation of the five-membered ring, and can be related to the RCP of QTAIM. These two types of interactions encompassing a single volume in isosurface suggest a weak IHB in this conformer, explaining the fact of not having been detected by QTAIM. Moreover, the peak at negative sign (λ2)ρ(r) (−0.018 au) in Fig. 7c indicates also the presence of an IHB. In accordance with the QTAIM, the NCI analysis shows no IHB in conformers trans-IIIb and trans-Vc.
 | ||
| Fig. 7 (a) QTAIM molecular graphs, (b) NCI isosurfaces generated with s = 0.5 au and blue-green-red scaling from −2 au < (λ2)ρ(r) < 2 au, and (c) NCI plots of the reduced density gradients [s(r)] versus sign (λ2)ρ(r) for the AcAspOMe conformers, obtained at the ωB97X-D/aug-cc-pVTZ level of theory. | ||
The contribution of the steric repulsion and hyperconjugative effects in the conformational preferences of the studied compound were evaluated through the NBO analysis. According to the data shown in Table 6, it appears that, as occurred for the AspOMe, a balance between these effects is crucial to the order of stability. Although the conformer trans-IIIb is the one that suffers minor steric repulsion (Erel,Lewis), it is also the geometry less stabilized by hiperconjugations (Erel,Hyper). The opposite is valid for trans-Ia and trans-Vc. Therefore, it was verified that the conformer trans-Ia has the resulting more favorable energetic balance in isolated phase, giving its greater stability. Furthermore, the severe increase of steric repulsion in conformers of AcAspOMe compared to the AspOMe was remarkable from NBO analysis.
| trans-Ia | trans-IIIb | trans-Vc | |
|---|---|---|---|
| a Relative energies in kcal mol−1. | |||
| Erel,Tot | 0.00 | 3.81 | 3.59 |
| Erel,Lewis | 16.18 | 0.00 | 16.25 |
| Erel,Hyper | 20.00 | 0.00 | 16.47 |
In order to assess the conformational variations induced by solvent effects, the AcAspOMe 1H NMR parameters were recorded in several aprotic solvents (Table 7). There are meaningful 3JHaHb value variations with the change of solvent, suggesting that the conformational populations are also varied. Indeed, when solvents were included in the calculations (Table 8), changes in the populations were noted. These variations are in accordance with the dipole moment of each conformer, since those with the highest values (trans-IIIb and trans-Vc) are the most stabilized in more polar solvents. At the same time, the trans-Ia, with the lowest dipole moment, was destabilized as the solvent becomes more polar. Although such destabilization, the conformer trans-Ia remains as the most stable, even in DMSO (about 72%), resulting from a more favorable energy balance due to the joint effect of stabilizing (hyperconjugation) and destabilizing (steric effects) energies (Table S6†).
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Solvent | εb | δNH | δHa | δHb1 | δHb2 | δHc | δHd | δHe | 3JHaHb1 | 3JHaHb2 | 3JHaH(N) |
| a Error in the measurements of J = ±0.05 Hz.b Dielectric constant. | |||||||||||
| C6D6 | 2.3 | 6.28 | 4.90 | 2.80 | 2.72 | 3.28 | 3.21 | 1.49 | 4.5b | 4.7 | 8.1 |
| CDCl3 | 4.8 | 6.51 | 4.86 | 3.04 | 2.86 | 3.77 | 3.70 | 2.04 | 4.3 | 4.5 | 7.9 |
| CD2Cl2 | 9.1 | 6.45 | 4.81 | 2.98 | 2.82 | 3.73 | 3.67 | 1.98 | 4.6 | 4.6 | 8.1 |
| Acetone-d6 | 20.7 | 7.43 | 4.79 | 2.84 | 2.81 | 3.67 | 3.63 | 1.92 | 5.9 | 5.9 | 8.1 |
| CD3CN | 37.5 | 6.84 | 4.71 | 2.80 | 2.77 | 3.67 | 3.64 | 1.89 | 6.0 | 5.6 | 8.1 |
| DMSO-d6 | 46.7 | 8.37 | 4.61 | 2.78 | 2.69 | 3.62 | 3.61 | 1.83 | 6.0 | 7.3 | 7.8 |

| Conformer | μ | Benzene | CCl4 | Chloroform | Dichloromethane | Acetone | Acetonitrile | DMSO | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Erel | P | Erel | P | Erel | P | Erel | P | Erel | P | Erel | P | Erel | P | ||
| a ZPE correction was included. | |||||||||||||||
| trans-Ia | 2.66 | 0.00 | 97.7 | 0.00 | 97.8 | 0.00 | 94.1 | 0.00 | 88.3 | 0.00 | 84.5 | 0.00 | 82.3 | 0.00 | 80.9 |
| trans-IIIb | 4.24 | 2.61 | 1.2 | 2.63 | 1.2 | 1.97 | 3.4 | 1.49 | 7.1 | 1.34 | 8.8 | 1.24 | 10.1 | 1.18 | 11.0 |
| trans-Vc | 3.47 | 2.65 | 1.1 | 2.66 | 1.0 | 2.14 | 2.5 | 1.75 | 4.6 | 1.50 | 6.7 | 1.41 | 7.6 | 1.37 | 8.1 |
Besides 3JHaHb, the coupling constant 3JHH H–N–Cα–H can be observed in spectra, which provides important information on the value of the dihedral angle between these atoms. However, the experimental value of 3JHaH(N) (Table 7) is practically constant with the change of the medium, which was expected, since the three conformers have similar H–N–Cα–H dihedral angles. Although these results are not able to indicate the predominance of one conformer, they confirm the agreement between experimental and theoretical data.
The carbonyl region of the infrared spectra for AcAspOMe is shown in Fig. 8. The experimental curve was deconvoluted in two bands, verifying the larger predominance of a component in all solvents, which was attributed to the conformer trans-Ia. The minor component, correspondent to trans-IIIb and trans-Vc, shows a progressive stabilization as the solvent polarity increases (from 7.0% in CCl4 to 20.9% in CH3CN). These experimental results presented an excellent agreement with the predicted ones by theoretical calculations, confirming the higher stability of trans-Ia.
 | ||
| Fig. 8 IR spectra of AcAspOMe showing the analytically resolved carbonyl stretching band in: (a) carbon tetrachloride, (b) chloroform, (c) dichloromethane and (d) acetonitrile. | ||
Analogously to the AspOMe, the calculated 3JHaHb for each conformer of the AcAspOMe (Fig. S4a in the ESI†) are invariable with the solvent exchange. However, as shown in Fig. S4b (in the ESI†), the experimental variations of 3JHaHb result from the contribution values (ηix3JHaHb,i) of each conformer. In nonpolar solvents, the conformer trans-Ia presents a conformational contribution much larger than the other two geometries. Since its calculated 3JHaHb values are about 4.0 Hz (Fig. S4a†), the corresponding experimental values listed in Table 7 (about 4.5 Hz) can be essentially attributed to this geometry. In turn, the increase of the experimental 3JHaHb in more polar solvents (i.e. 3JHaHb1 = 6.0 and 3JHaHb2 = 7.3 Hz, in DMSO) can be explained by the stabilization of forms b and c. It is worth noting that the higher variations in the 3JHaHb for the AcAspOMe, compared to the AspOMe, corroborate with the fact that their conformers, mainly the trans-Ia, present more significant changes in their populations in solution. Thus, the experimental 1H NMR data showed excellent concordance with the theoretical calculations, enabling the examination of the conformational equilibrium of the AcAspOMe including the solvent effects.
4. Conclusions
A joint application of experimental and theoretical data allowed us to make predictions about the conformational preferences of AspOMe and AcAspOMe. The experimental and calculated 3JHH couplings constants were found to be in good agreement and provided insights into the conformational behavior of the compounds in solution. The same is valid with respect to the IR data for AcAspOMe. In turn, the theoretical results can explain the experimental ones, since they were used to determine the most stable conformers and their relative energies, both in isolated phase and solution. They also indicated that inclusion of implicit solvent model (IEF-PCM) leads to small changes in conformer populations related to the isolated phase, but occurs the prevalence of the Ia and IIa conformers for AspOMe and trans-Ia for AcAspOMe.NBO, QTAIM and NCI analysis showed that although the presence of an IHB has been observed in some conformers of these compounds, both steric and hyperconjugative effects have remarkable importance in their conformational preferences, as in isolated phase as in solution.
Thus, to explore the conformational preferences of amino acids derivatives and to map the more significant interactions that are present in these systems are essential for the understanding of more complex molecules of great biological importance.
Acknowledgements
The authors thank a grant #2012/03933-5 from São Paulo Research Foundation (FAPESP) for financial support of this work and for a scholarship (to C.B.B. #2012/18567-4 and L.C.D). Thanks also to Conselho Nacional de Pesquisa (CNPQ) for a fellowship (to R.R.).Notes and references
- M. E. Sanz, J. C. López and J. L. Alonso, Phys. Chem. Chem. Phys., 2010, 12, 3573 RSC.
- R. M. Balabin, Comput. Theor. Chem., 2011, 965, 15 CrossRef CAS PubMed.
- M. M. Quesada-Moreno, A. A. Márquez-García, J. R. Avilés-Moreno and J. J. López-Gonzáles, Tetrahedron: Asymmetry, 2013, 24, 1537 CrossRef CAS PubMed.
- M. De Poli, M. De Zotti, J. Raftery, J. A. Aguilar, G. A. Morris and J. Clayden, J. Org. Chem., 2013, 78, 2248 CrossRef CAS PubMed.
- M. De Zotti, F. Formaggio, M. Crisma, C. Peggion, A. Moretto and C. Toniolo, J. Pept. Sci., 2014, 20, 307 CrossRef CAS PubMed.
- L. J. Klasic, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 161 CrossRef.
- M. Chen and Z. Lin, J. Phys. Chem., 2007, 127, 154314 CrossRef PubMed.
- T. Kimura, N. Matubayasi, H. Sato, F. Hirata and M. Nakahara, J. Phys. Chem. B, 2002, 106, 12336 CrossRef CAS.
- R. A. Cormanich, L. C. Ducati, C. F. Tormena and R. Rittner, J. Phys. Org. Chem., 2013, 26, 849 CrossRef CAS.
- C. B. Braga, L. C. Ducati, C. F. Tormena and R. Rittner, J. Phys. Chem. A, 2014, 118, 1748 CrossRef CAS PubMed.
- R. A. Cormanich, L. C. Ducati, C. F. Tormena and R. Rittner, Spectrochim. Acta, Part A, 2014, 123, 482 CrossRef CAS PubMed.
- R. A. Cormanich, L. C. Ducati and R. Rittner, Chem. Phys., 2011, 387, 85 CrossRef CAS PubMed.
- R. A. Cormanich, L. C. Ducati and R. Rittner, J. Mol. Struct., 2012, 1014, 12 CrossRef CAS PubMed.
- R. A. Cormanich, L. C. Ducati, C. F. Tormena and R. Rittner, Chem. Phys., 2013, 421, 32 CrossRef CAS PubMed.
- N. Narendra, T. M. Vishwanatha and V. V. Sureshbabu, Synthesis, 2011, 3247 CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS PubMed.
- Y. Zhao, N. E. Schultz and D. G. J. Truhlar, J. Chem. Theory Comput., 2006, 2, 364 CrossRef.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- S. J. Grimme, Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef PubMed.
- V. Barone, J. Chem. Phys., 1994, 101, 6834 CrossRef PubMed.
- G. F. Gauze, E. A. Basso, R. H. Contreras and C. F. Tormena, J. Phys. Chem. A, 2009, 113, 2647 CrossRef CAS PubMed.
- NBO, version 5.0, implemented in the Gaussian 09 package of programs, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Wallingford, Gaussian, Inc., 2009 Search PubMed.
- T. A. Keith, AIMALL (version 11.12.19), TK Gristmill Software, Overland Park, KS, USA, 2011, http://aim.tkgristmill.com Search PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. J. Yang, Chem. Theory Comput., 2011, 7, 625 CrossRef PubMed.
- R. F. W. Bader, Chem.–Eur. J., 2006, 12, 2896 CrossRef CAS PubMed.
- U. Koch and P. L. A. Popelier, J. Phys. Chem., 1995, 99, 9747 CrossRef CAS.
- J. R. Lane, J. Contreras-García, J.-P. Piquemal, B. J. Miller and H. G. Kjaergaard, J. Chem. Theory Comput., 2013, 9, 3263 CrossRef CAS; D. C. Solha, T. M. Barbosa, R. V. Viesser, R. Rittner and C. F. Tormena, J. Phys. Chem. A, 2014, 118, 2794 CrossRef PubMed.
- M. Karplus, J. Chem. Phys., 1959, 30, 11 CrossRef CAS PubMed.
- J. Poater, J. J. Dannenberg, M. Solà and F. M. Bickelhaupt, Advances in Chemical Modeling, M. V. Putz, Nova Science, Hauppauge, NY, 2011, pp. 139–153 Search PubMed.
- R. M. Balabin, J. Chem. Phys., 2010, 132, 211103 CrossRef PubMed.
- T. van Mourik, J. Comput. Chem., 2007, 29, 1 CrossRef PubMed.
- L. F. Holroyd and T. van Mourik, Chem. Phys. Lett., 2007, 442, 42 CrossRef CAS PubMed.
- N. Y. Palermo, J. Csontos, M. C. Owen, R. F. Murfhy and S. Lovas, J. Comput. Chem., 2007, 28, 1208 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and theoretical evaluation on the conformational behavior of L-aspartic acid dimethyl ester and its N-acetylated derivative. See DOI: 10.1039/c4ra14480e |
| This journal is © The Royal Society of Chemistry 2015 |