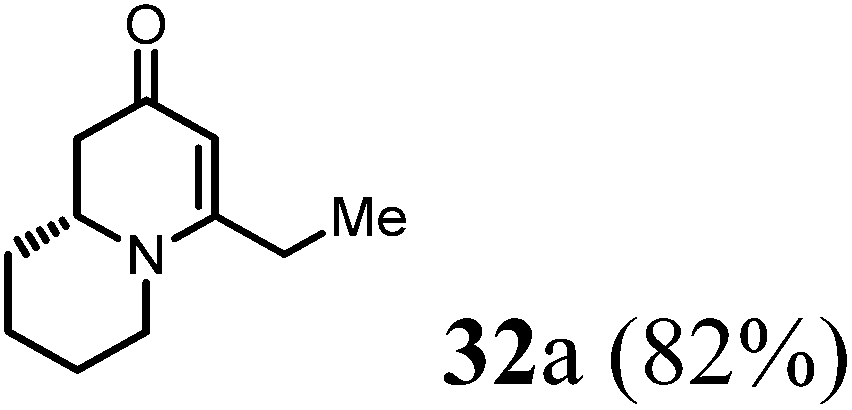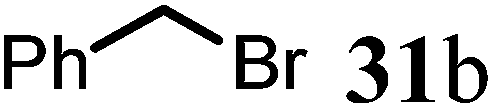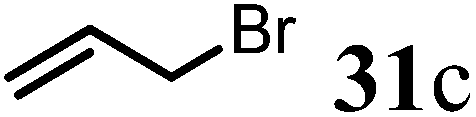DOI:
10.1039/C4RA14418J
(Paper)
RSC Adv., 2015,
5, 18894-18908
Building polyfunctional piperidines: a stereoselective strategy of a three-component Mannich reaction inspired by biosynthesis and applications in the synthesis of natural alkaloids (+)-241D; (−)-241D; isosolenopsin A and (−)-epimyrtine†
Received
12th November 2014
, Accepted 6th February 2015
First published on 6th February 2015
Abstract
A general method to assemble multi-substituted chiral piperidines was developed, inspired by the biosynthesis of piperidine natural products. In biosynthesis, Δ1-piperideine 4 plays a key role as a common intermediate giving rise to a variety of piperidine-based natural alkaloids. Nature uses L-lysine as a building block, enzymatically transforming it into a δ-amino carbonyl intermediate 3 as the precursor to cyclize into Δ1-piperideine 4. We envisioned that such a process could be accomplished by a vinylogous type Mannich reaction if a functionalized dienolate was employed. A stereoselective three-component vinylogous Mannich-type reaction (VMR) of 1,3-bis-trimethylsily enol ether 7 was therefore investigated and was found to give cyclized chiral dihydropyridinone compound 9 as an adduct. Like Δ1-piperideine in biosynthesis, the chiral 2,3-dihydropyridinone compound 9 from VMR is a versatile intermediate for building a variety of new chiral piperidine compounds. The method was showcased by concise two-step approaches in the synthesis of the bioactive natural alkaloids (+)-241D; (−)-241D and isosolenopsin A. Furthermore, when properly functionalized substrate aldehyde 24 was employed, the corresponding dihydropyridinone adduct 25 cyclized to form a second piperidine ring, leading to a chiral polyfunctional quinolizidine enaminone 27. This versatile intermediate was used to prepare a variety of new chiral quinolizidine compounds, including natural alkaloid (−)-epimyrtine.
Introduction
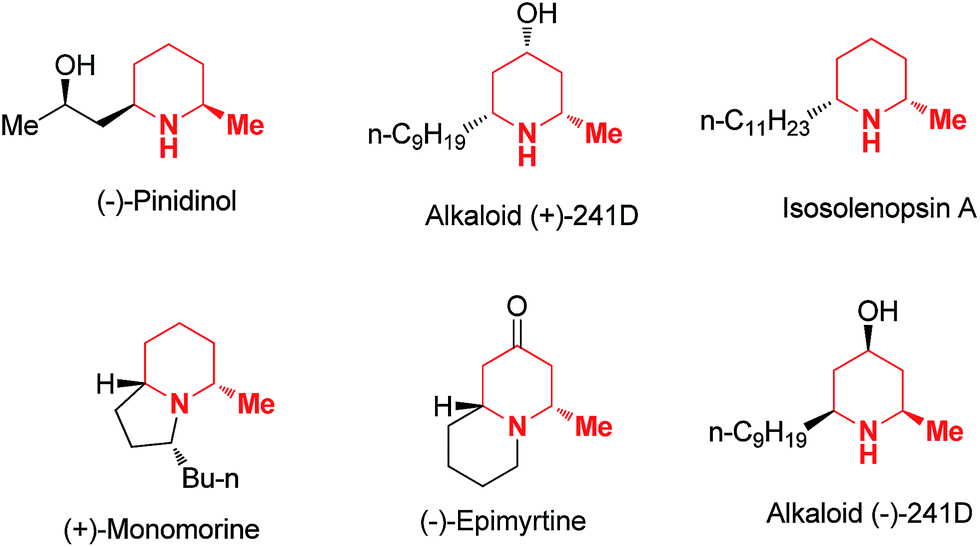
Functionalized piperidine rings are common moieties incorporated in a variety of natural alkaloids and pharmaceutical molecules.1 In fact, piperidine is the most frequently used non-aromatic ring in small molecule drugs listed in the FDA orange book.2 Developing synthetic approaches for the stereoselective construction of these ring systems has been an area of intense research in synthetic organic chemistry for decades.3 Among the various piperidine derivatives, 2 and/or 6 substituted piperidines are particularly common and interesting4 since such substitution patterns block the metabolism of the piperidine ring and potentially have a significant impact on the ring's 3D conformation. For such reasons, installation of α substitutions adjacent to the piperidine nitrogen are commonly employed as a strategy in medicinal chemistry research to tune either biological activities or pharmacological properties. In practice, the methyl group is one of the most common and simplest substituents serving this purpose. Interestingly, α-methyl multi-substituted piperidines are also commonly found in naturally occurring piperidine alkaloids such as (−)-pinidinol, (+)-241D and isosolenopsin A etc. (Fig. 1). Some of these natural alkaloids have demonstrated interesting pharmacological properties and served as valuable starting points for new drug discovery.5
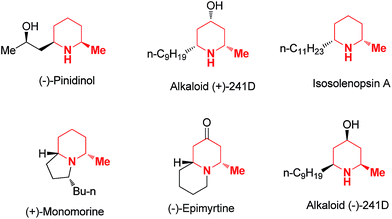 |
| | Fig. 1 Examples of natural alkaloids incorporating α-methyl substituted piperidines. | |
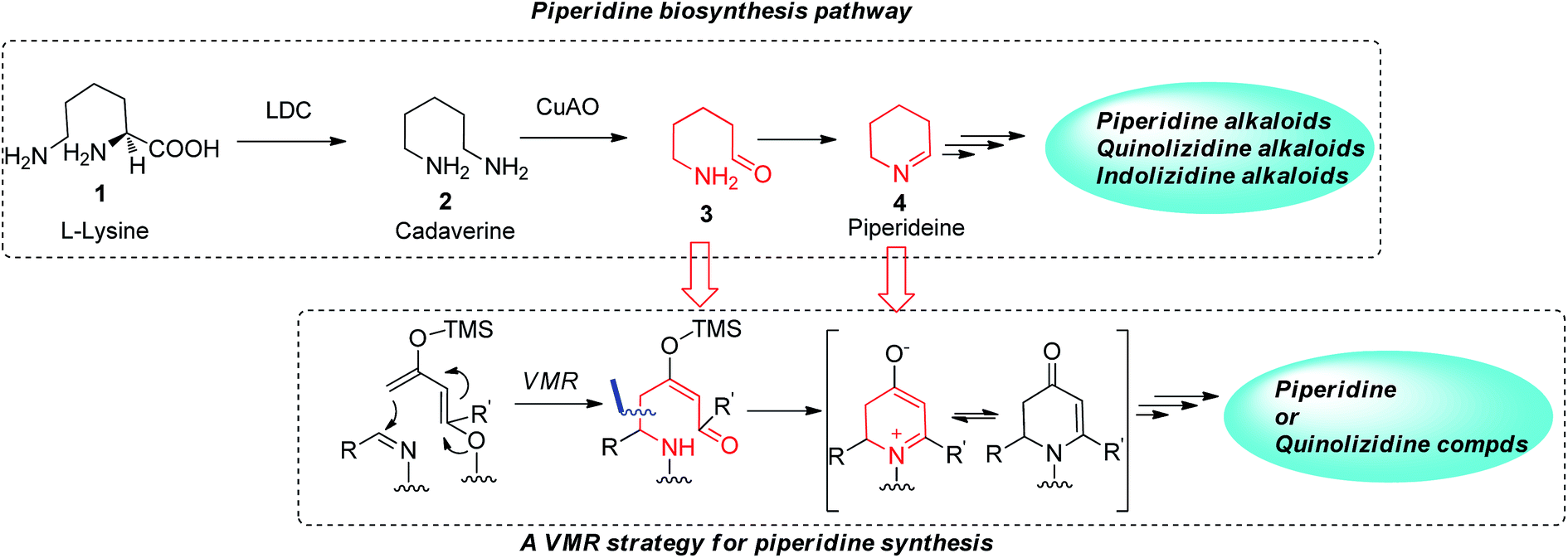
The biosynthetic pathway of many piperidine-based natural alkaloids has been studied. Δ1-Piperideine 4, which forms from an intramolecular imine cyclization of a δ-amino pentanal precursor 3, was believed to be a key common intermediate in the pathway. Studies have shown that further transformations on this prototype piperidine ring lead to a variety of structurally diversified piperidine, quinolizidine and indolizidine alkaloids in nature.6 The basic starting building block in this pathway is L-lysine, which undergoes several enzymatically catalyzed transformations, including decarboxylation by LDC (lysine decarboxylase) and oxidative deamination by CuAO (copper amine oxidase). The resulting δ-amino pentanal 3 then gives rise to the key Δ1-piperideine ring (Fig. 2). However, without nature's powerful enzyme tools, chemical synthesis of Δ1-piperideine is tedious7 due to its instability and such intermediate is therefore not practical to be widely applied in synthesis lab like its role in biosynthesis.8 We envisioned however that similar δ-amino carbonyl precursor for Δ1-piperideine can be assembled conveniently via a vinylogous Mannich-type reaction (VMR) with an aldimine if a properly functionalized dienolate was employed. As shown in Fig. 2, cyclization of the initial δ-amino carbonyl adduct would lead to a 2,3-dihydropyridinone, which could also be viewed as a tautomeric form of cyclic imine, but more stable and easier to handle (Fig. 2). In fact, the synthetic utility of dihydropyridinones has been extensively investigated by the Comins group, but to date the methodology for preparation of these intermediates has been limited.9 Here we report the successful implementation of the VMR strategy to generate useful chiral dihydropyridone intermediates, and their subsequent transformation to a variety of interesting piperidine-containing natural products and compounds of medicinal interest.
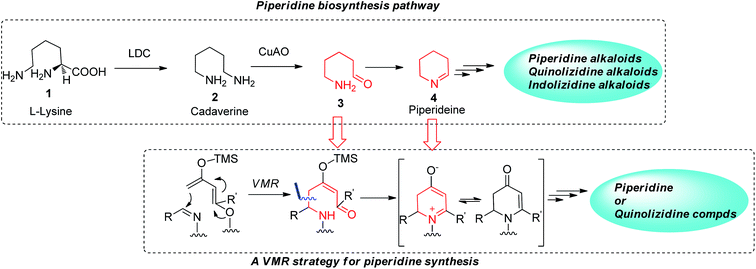 |
| | Fig. 2 VMR strategy for piperidine synthesis inspired by biosynthesis. | |
Results and discussion
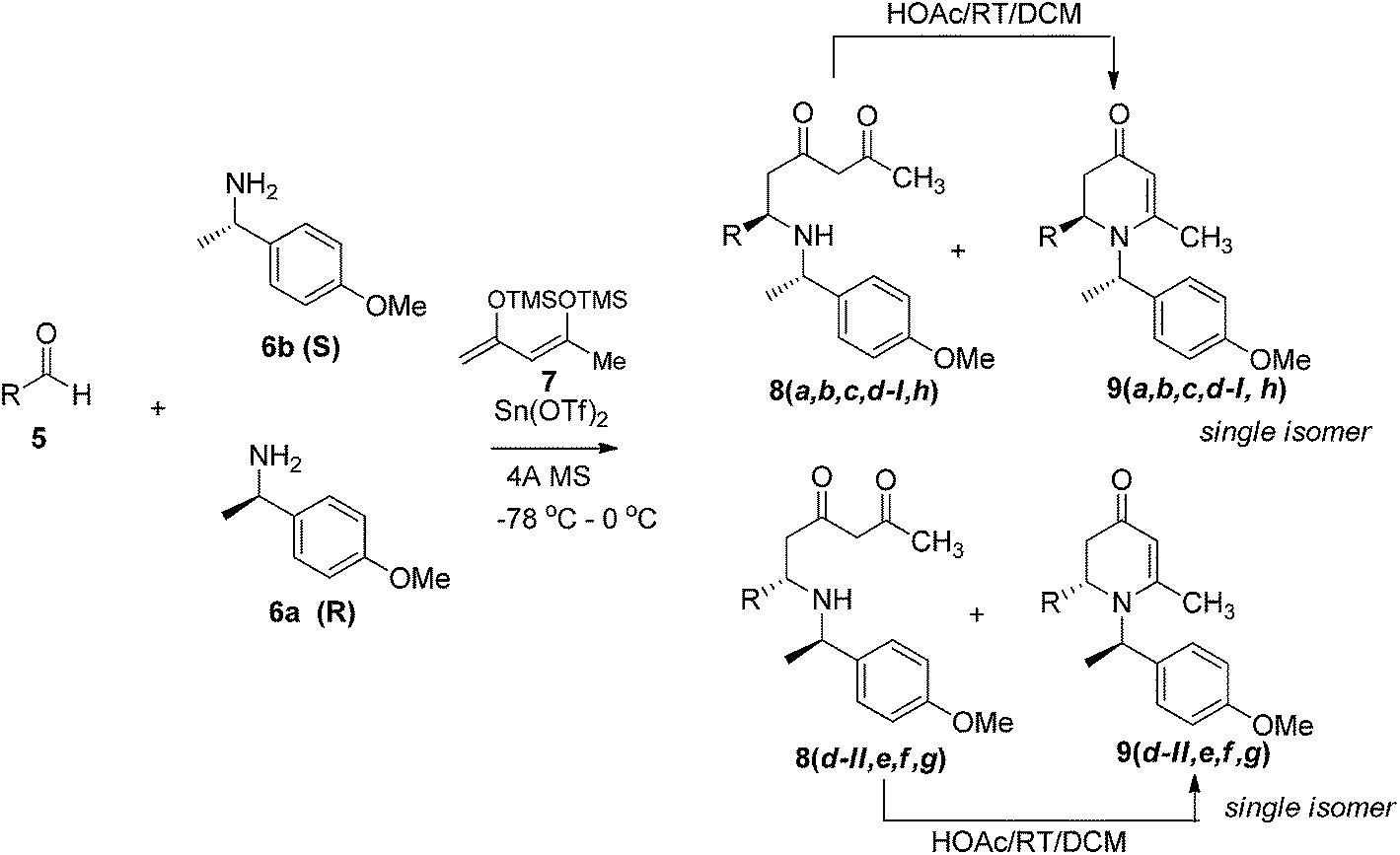
The simple 1,3-bis-trimethylsily enol ether 7 has been employed as a vinylogous nucleophilic reagent in several organic transformations such as cyclization with 1,2-dielectrophiles, bromination, and vinylogous aldol reaction.10 Surprisingly, the use of 7 as dienolate in a Mannich-type reaction has never been reported.11 To ensure stereoselective control in VMR, inexpensive commercially available chiral α-methyl benzylamine 6 was employed to form chiral aldimines in situ. The three-component VMR reaction of 6 and 7 with various aldehydes 5 was carried out in the presence of Sn(OTf)2 in DCM at −78 °C to 0 °C. Corresponding adducts 8 were observed from reaction LC-MS analysis, however in a mixture with cyclized 2,3-dihydropyridinone products 9. Treatment of the crude mixture with a catalytic amount of acetic acid in DCM led to complete conversion of acylic adducts 8 into 9 (Scheme 1).
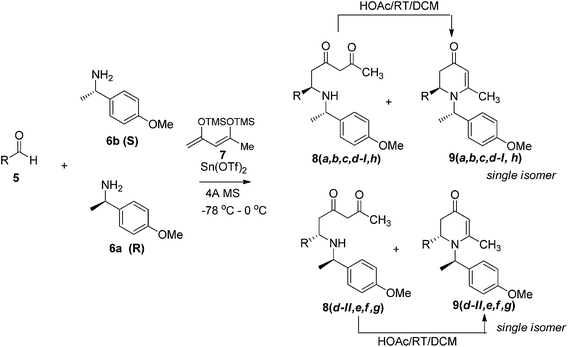 |
| | Scheme 1 | |
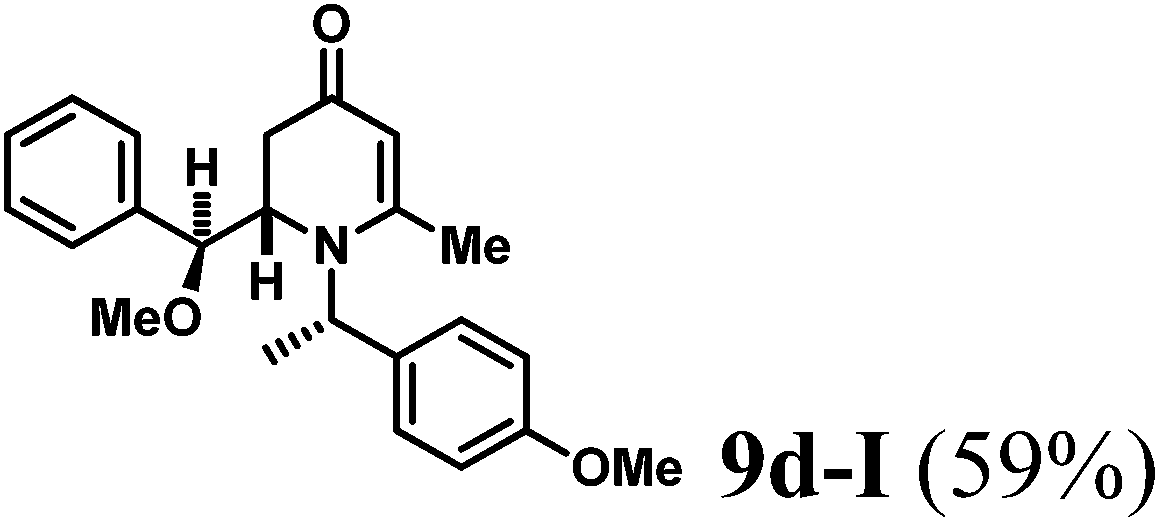
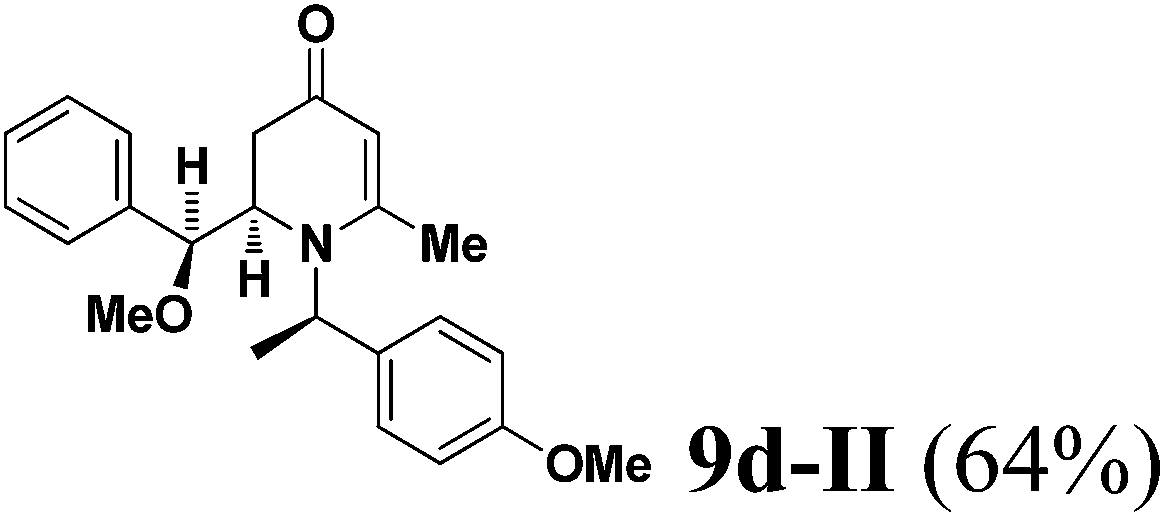
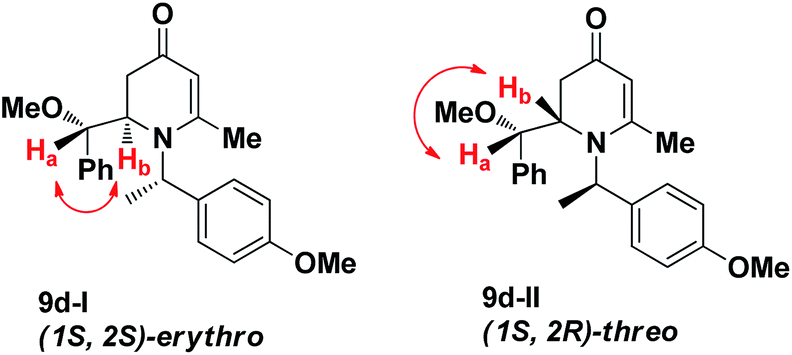
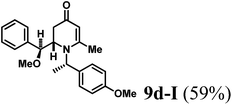
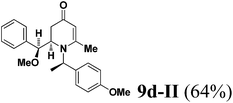
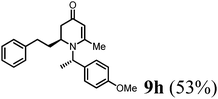
The results of the VMR reaction of 7 with various aldehydes are summarized in Table 1. Most of the reactions showed moderate to good yields. A variety of functional groups were well tolerated. The reactions showed excellent diastereoselectivities since in all cases only single isomers were observed and isolated from the reaction mixtures. In order to confirm that the stereoselectivities of the reaction were auxiliary directed, compounds 9d-I and 9d-II were prepared from the same chiral substrate aldehyde 5d, in the presence of chiral amine auxiliary 6a and its enantiomer 6b. The proton NMR spectra of these compounds showed that the JHa/Hb value for 9d-I was 8.80 Hz while the corresponding value for 9d-II was 9.2 Hz, suggesting that 9d-I and 9d-II were the erythro and threo isomers respectively, based on literature precedent.12 These results confirmed auxiliary directed stereoselectivities and further supported the established sense of stereochemical induction in such VMR13 (Fig. 3).
Table 1 Asymmetric three-component vinylogous Mannich reactions of 1,3-bis-trimethylsily enol ether 7
| Entry |
Substrate 5 |
6
|
9 (yield) |
| 1 |
|
6b
|
|
| 2 |

|
6b
|
|
| 3 |

|
6b
|
|
| 4 |
|
6b
|
|
| 5 |
|
6a
|
|
| 6 |
|
6a
|
|
| 7 |
|
6a
|
|
| 8 |
|
6a
|
|
| 9 |
|
6b
|
|
 |
| | Fig. 3 NMR analysis of 9d-I and 9d-II. | |
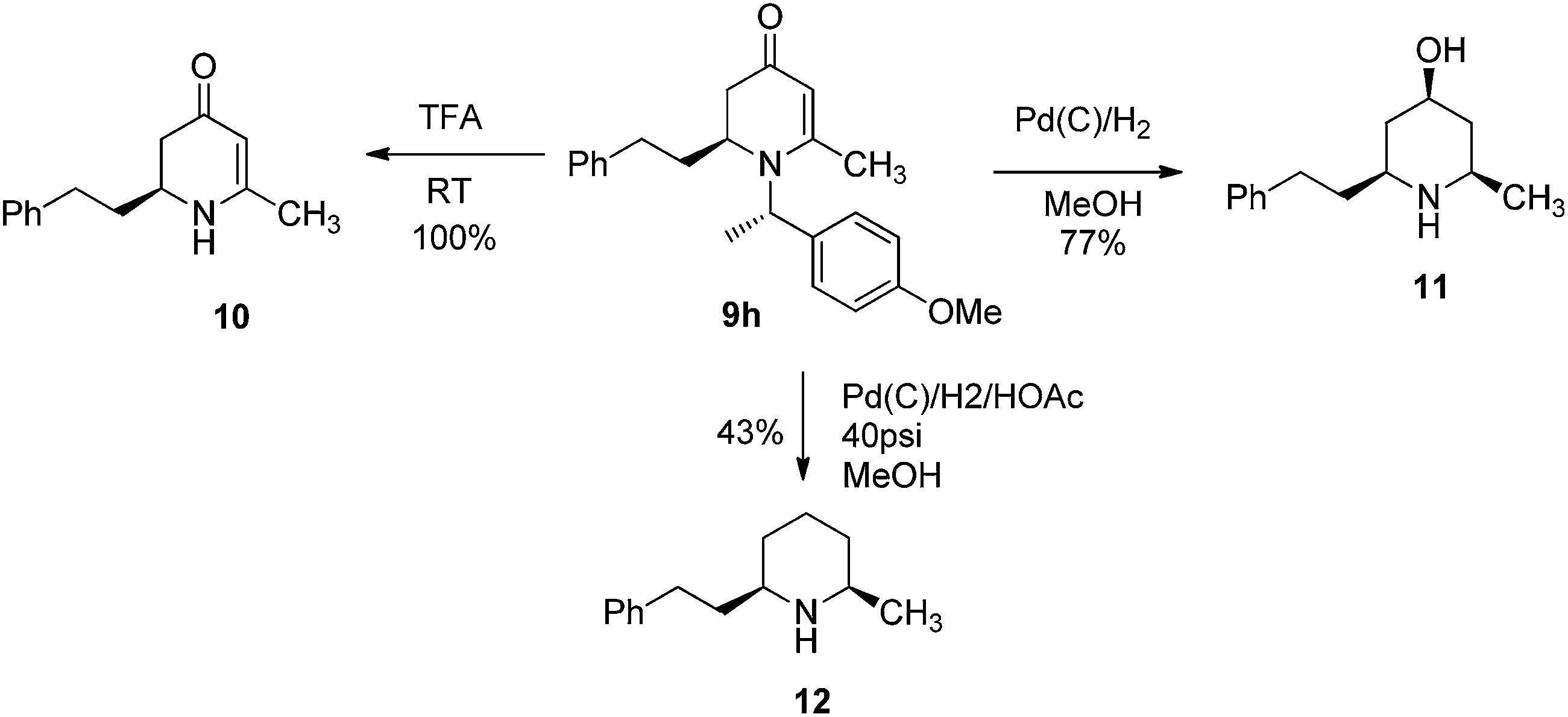
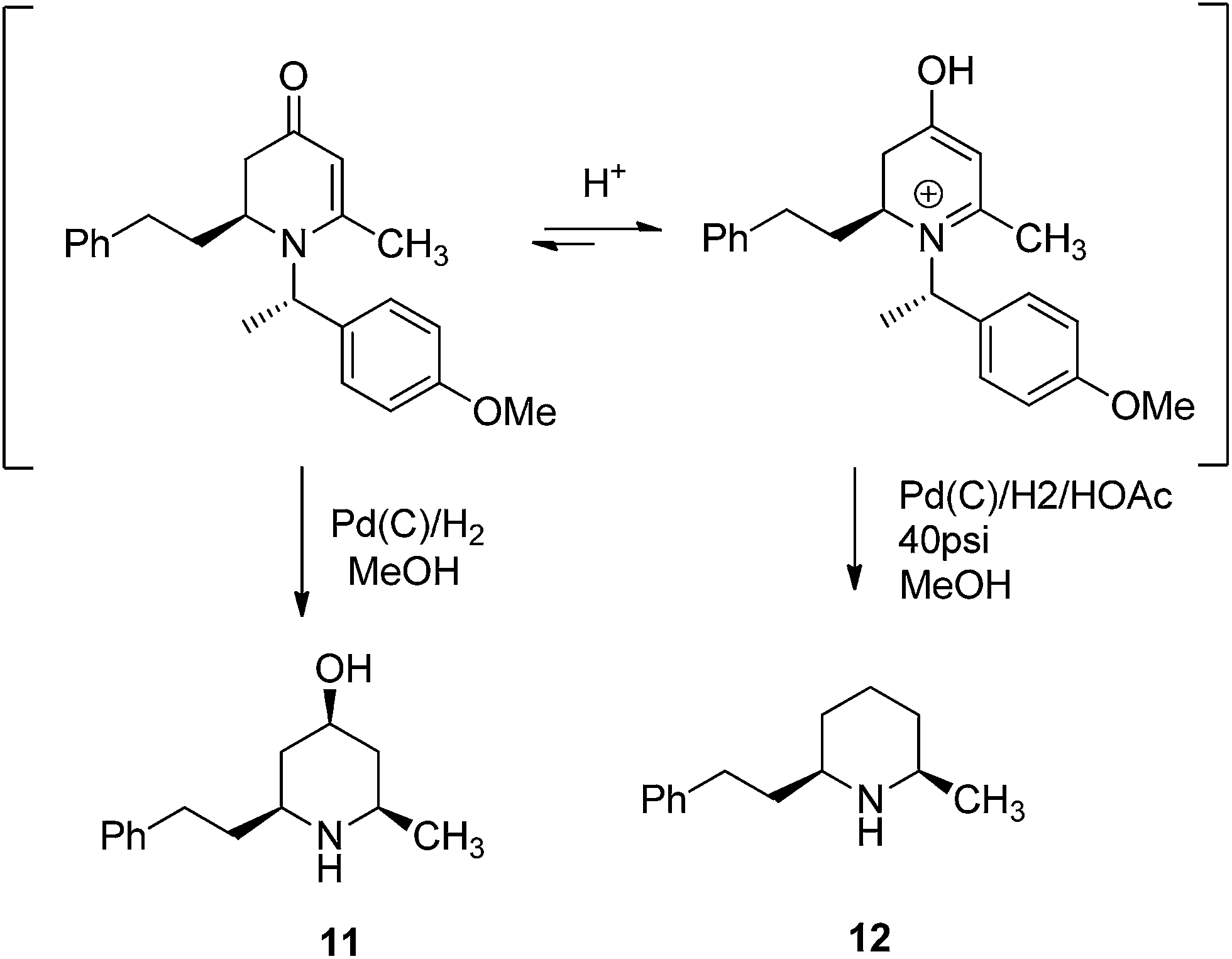
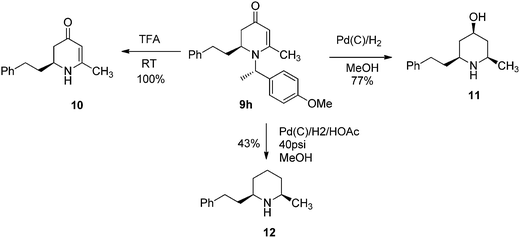
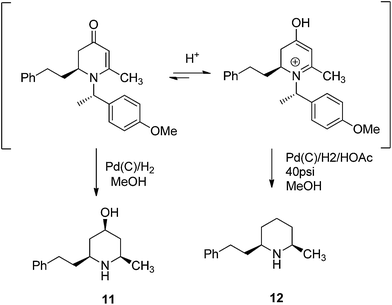
To examine the synthetic utility of 2,3-dihydropyridinones obtained from the VMR, adduct compound 9h was selected to probe further transformations. When the compound 9h was treated with TFA at room temperature, the chiral benzyl directing group was cleaved to give cyclic enaminone 10 in quantitative yield (Scheme 2). We also found that the corresponding chiral substituted piperidine could be obtained from 9hvia palladium catalyzed hydrogenation. Interestingly, under different hydrogenation conditions, the reduction of 9h yielded different major piperidine products. When hydrogenation was performed in MeOH in presence of palladium on carbon at room temperature, the reaction cleaved the chiral benzyl group and saturated the 2,3-dihydro-4-pyridinone simultaneously to give cis-3-hydroxy 2,6-disubstituted piperidine compound 11 stereospecifically as the major product, accompanied by deoxygenated piperidine compound 12 as the minor product (11/12, ratio 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)14 (Scheme 2). However, when the hydrogenation was performed in a Parr hydrogenator under 40 psi hydrogen pressure in a mixture of methanol and acetic acid (1/1), the major product was deoxygenated cis-2,6-dialkylated piperidine 12 accompanied by 11 as the minor product (12/11, 5:1) (Scheme 2). The results could be explained by a shift in the equilibrium between 2,3-dihydropridinone and 2,3-dihydropridinium under different conditions.15 Presumably, 2,3-dihydropridinone is the major species present under neutral conditions and hydrogenation led to 4-hydroxy piperidine product 11. However, under acidic conditions, the protonated 2,3-dihydropridinium species is the major (or more reactive) species present, and hydrogenation gives the corresponding deoxygenated piperidine 12 as the major product16 (Fig. 4). Similar hydrogenations of 2,3-dihydropyridinones have been reported to yield 4-hydroxy piperidine compounds stereospecifically.14 However, to our knowledge the direct deoxygenative reduction of 2,3-dihydropridinones is rarely reported. The results allowed accessing different substitution type piperidine compounds from 2,3-dihydropyridinones 9 by simply switching different reduction conditions.
:1)14 (Scheme 2). However, when the hydrogenation was performed in a Parr hydrogenator under 40 psi hydrogen pressure in a mixture of methanol and acetic acid (1/1), the major product was deoxygenated cis-2,6-dialkylated piperidine 12 accompanied by 11 as the minor product (12/11, 5:1) (Scheme 2). The results could be explained by a shift in the equilibrium between 2,3-dihydropridinone and 2,3-dihydropridinium under different conditions.15 Presumably, 2,3-dihydropridinone is the major species present under neutral conditions and hydrogenation led to 4-hydroxy piperidine product 11. However, under acidic conditions, the protonated 2,3-dihydropridinium species is the major (or more reactive) species present, and hydrogenation gives the corresponding deoxygenated piperidine 12 as the major product16 (Fig. 4). Similar hydrogenations of 2,3-dihydropyridinones have been reported to yield 4-hydroxy piperidine compounds stereospecifically.14 However, to our knowledge the direct deoxygenative reduction of 2,3-dihydropridinones is rarely reported. The results allowed accessing different substitution type piperidine compounds from 2,3-dihydropyridinones 9 by simply switching different reduction conditions.
 |
| | Scheme 2 | |
 |
| | Fig. 4 Equilibrium between 2,3-dihydropridinone and 2,3-dihydropridinium. | |
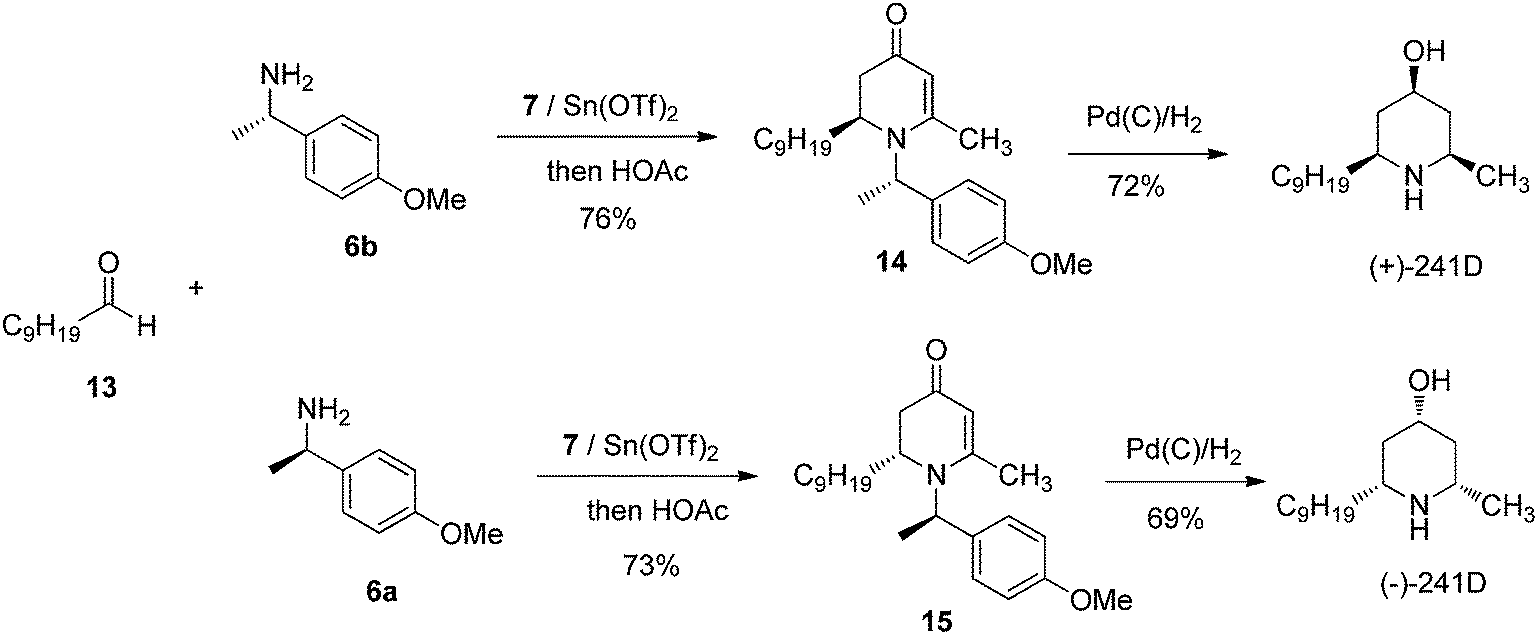
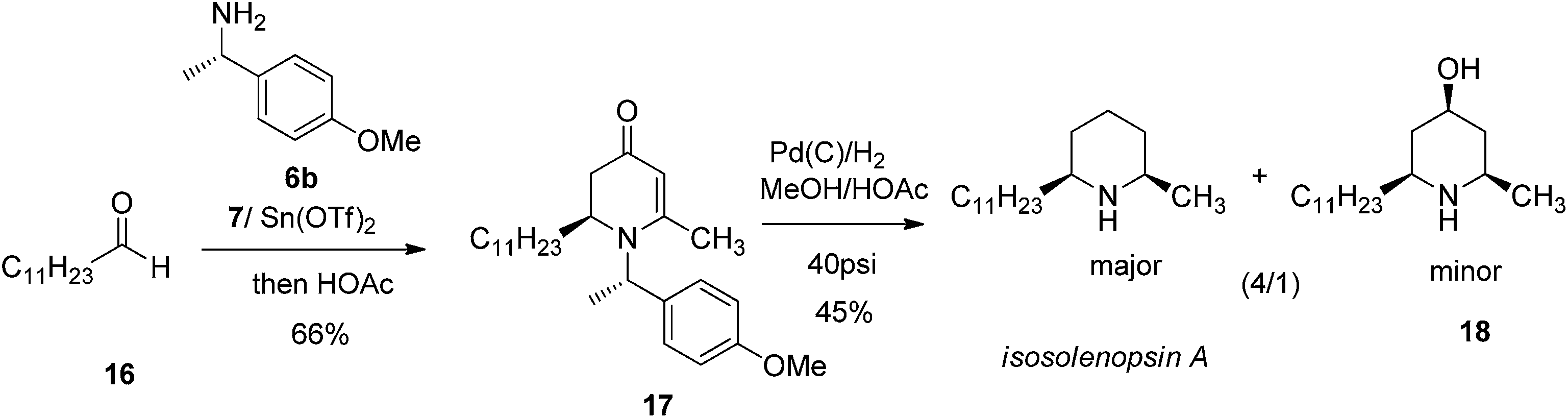
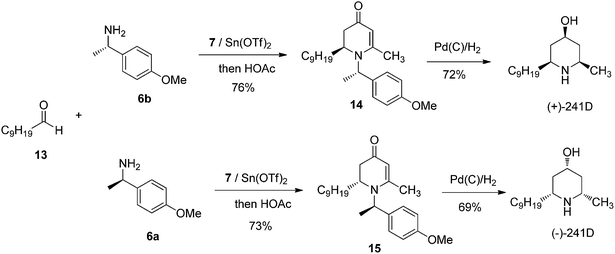
We further probed the utility of our chiral piperidine intermediates by applying the VMR methodology to the asymmetric synthesis of natural piperidine-containing alkaloids. Dendrobate alkaloid (+)-241D and its enantiomer (−)-241D were among our first targets. Dendrobate alkaloid (+)-241D was isolated from the methanolic skin extracts of the Panamanian poison frog Dendrobates speciosus.17 The alkaloid shows interesting bioactivity as a potent non-competitive blocker of acetylcholine and ganglionic nicotinic receptor channels.18 The structure of (+)-241D features an all-cis 2,4,6-trisubstituted piperidine core bearing three chiral centers. The asymmetric synthesis of (+)-241D has been reported by multiple research groups via a variety of synthesis routes employing between eight and eighteen steps.19 We were delighted to find that using the newly developed VMR strategy, the asymmetric synthesis of (+)-241D and its enantiomer could be accomplished simply in two steps from inexpensive commercial materials. Using chiral α-methyl benzylamines 6a & 6b to control stereochemistry, the reaction of bis-trimethylsily enol ether 7 with decanal 13 yielded chiral adducts 14 & 15 respectively. Subsequent reduction of 2,3-dihydro-4-pyridones 14 & 15 by palladium-catalyzed hydrogenation in methanol gave (+)-241D and (−)-241D in good yield (Scheme 3).
 |
| | Scheme 3 Enantioselective synthesis of (+)-241D and (−)-241D. | |
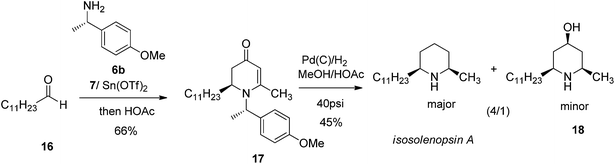
The versatile utility of such VMR approach in assembling piperidine was further exampled in asymmetric synthesis of another natural alkaloid isosolenopsin A which incorporate cis-2,6-dialkylpiperidine as a core. isosolenopsin A was isolated from the venom of the fire ant solenopsis and was found to have a variety of interesting bioactivities including antibiotic, antifungal, anti-HIV, blockade of neuromuscular transmission and potent and selective inhibition of the neuronal nitric oxide synthase.20 By the similar strategy, corresponding VMR adduct 2,3-dihydro-4-pyridones 17 was obtained when dodecanal 16 and chiral amine 6b were employed. The palladium-catalyzed reduction on 2,3-dihydro-4-pyridone 17 was carried out in methanol in presence of acetic acid (50%) under 40 psi hydrogen pressure in a Parr hydrogenator. Corresponding deoxygenated product isosolenopsin A was obtained as the major product in moderate yield (45%) (Scheme 4). The current approach presented the shortest route for asymmetric synthesis of isosolenopsin A than any other reported methods.21
 |
| | Scheme 4 Enantioselective synthesis of natural alkaloid isosolenopsin A. | |
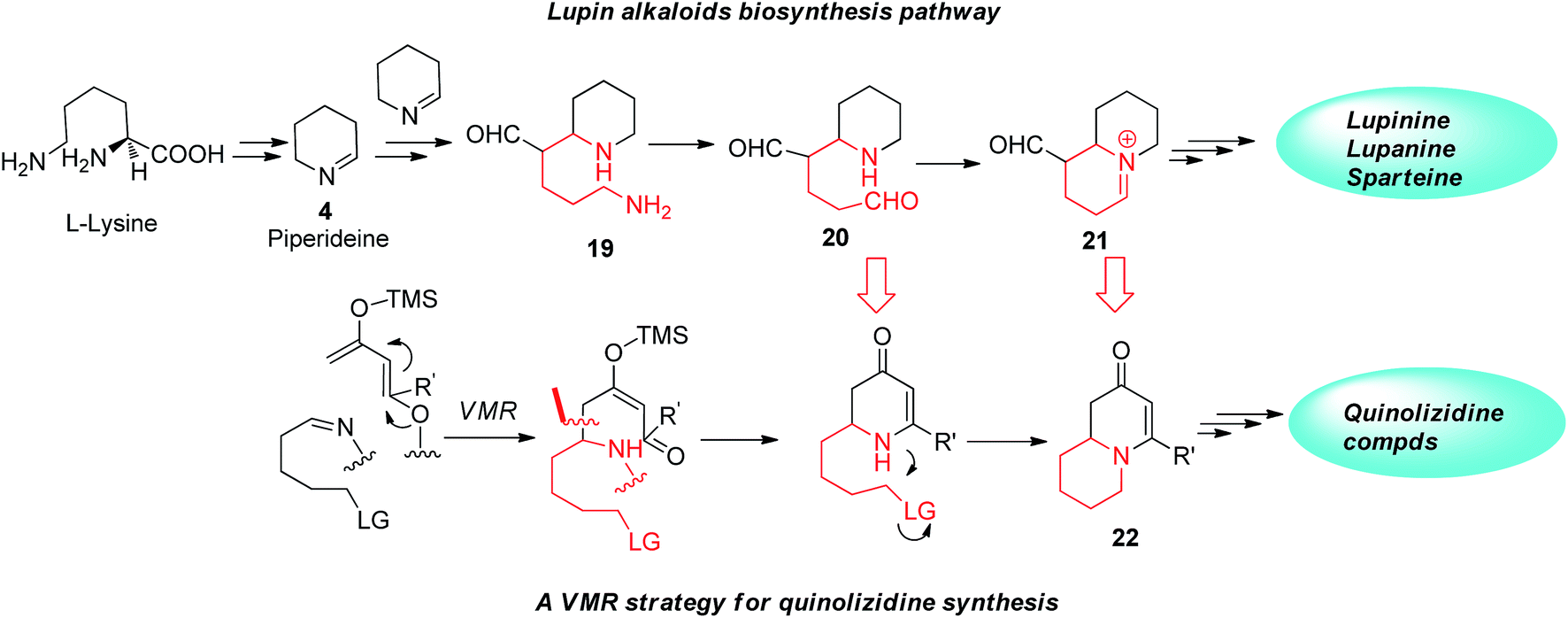
Beyond applying to building simple multi-substituted piperidine compounds, current VMR strategy also provides potentials in synthesizing chiral quinolizidine compounds. Quinolizidine compounds structurally incorporate two fused piperidine rings sharing common nitrogen. Like piperidine, quinolizidine represent both a class compound of pharmaceutical interest and an important family of natural alkaloids. In nature, several hundred structurally related quinolizidine compounds have been identified from a variety of natural sources, predominately from plants and amphibian skin.22 Some natural quinolizidine alkaloids exhibit interesting pharmacological properties23 and serve as important starting points for the drugs discovery.24 Interestingly, the biosynthesis of some quinolizidine alkaloids shares the same pathway of the natural piperidine alkaloids that undergo the same Δ1-piperideine intermediate 4, enzymatically starting from L-lysine.25 As an example, in the biosynthesis of quinolizidine alkaloids lupinine, Δ1-piperideine is also the key intermediate to assemble the first piperidine ring for the quinolizidine core. To build the second piperidine ring, two Δ1-piperideine intermediates undergo a cross aldol-type coupling and one of the imine systems gets hydrolyzed after coupling and undergoes oxidation resulting in primary amine function 20. Ultimately the formation of the quinolizidine nucleus in biosynthesis is accomplished by another intramolecular imine formation (Fig. 5). We however envisioned that in the VMR we developed, if the aldimine substrate has been properly functionalized, piperidine-like adducts arising from the asymmetric VMR may further conveniently cyclize to form second piperidine ring to give the desired quinolizidine product. As shown in Fig. 5, if a δ-leaving group is incorporated in the aldimine substrate and can be tolerated in the asymmetric VMR for the first piperidine ring construction, the quinolizidine structure 22 should be readily formed by a subsequent intramolecular SN2 cyclization (Fig. 5).
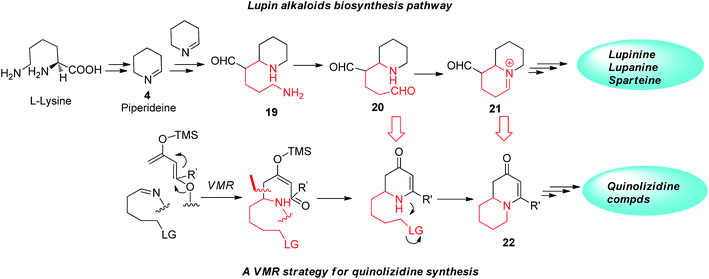 |
| | Fig. 5 VMR strategy for quinolizidine synthesis. | |
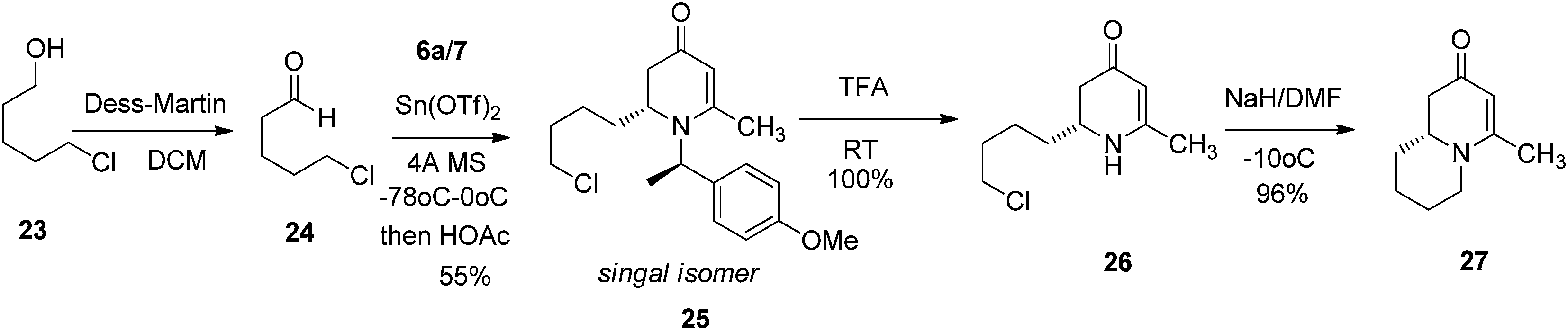
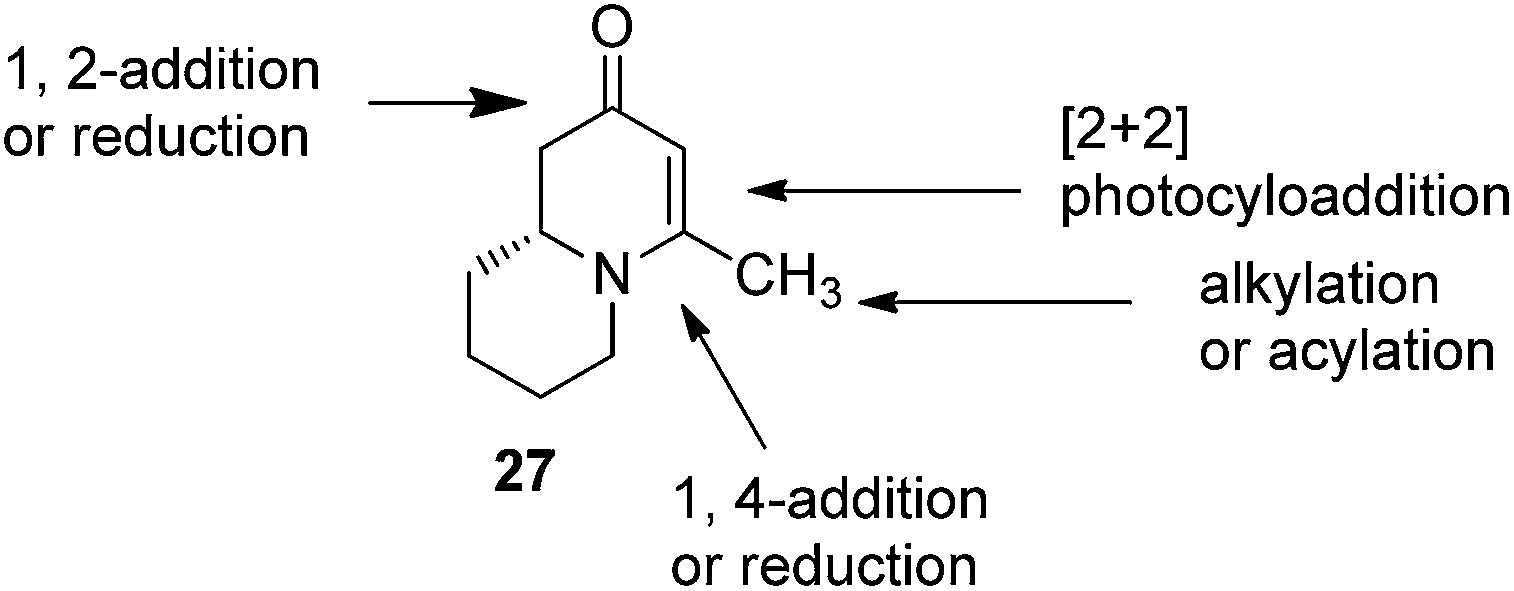
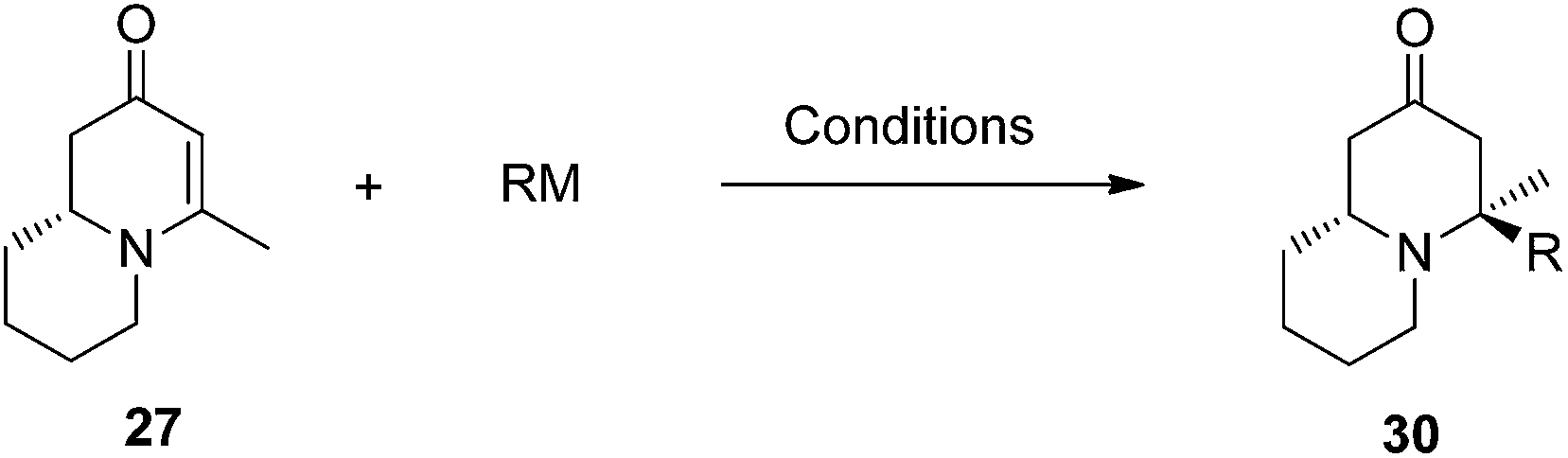
To test this idea 5-chloropentanal 24 was prepared from the oxidation of 5-chloropentan-1-ol 23 and the corresponding three-component VMR reaction was carried out. The reaction gave adduct 25 in expected excellent diastereoselectivity as a single stereoisomer. The δ chloride group which serve as a future leaving group on the substrate, was well tolerated (Scheme 5). With dihydropyridinone 25 in hand we set out to construct the second ring for a quinolizidine core. The α-methyl benzyl group was cleaved cleanly upon the treatment with TFA at room temperature overnight to give compound 26 in quantitative yield. In presence of sodium hydride in DMF, intramolecular SN2 cyclization by 26 led to a quinolizidine intermediate 27 as a cyclic enaminone (Scheme 5). We envisaged that such cyclic enaminone 27 could be a valuable polyfunctional quinolizidine intermediate since different organic transformations can be carried out at different positions on this molecule. It provides convenient entries to access different types chiral quinolizidine compounds (Fig. 6).
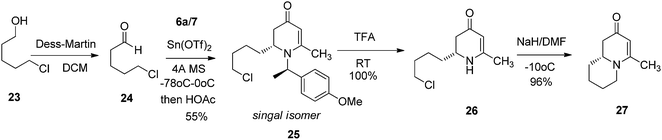 |
| | Scheme 5 | |
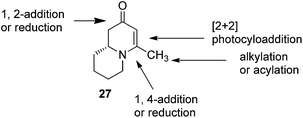 |
| | Fig. 6 Synthesis versatility of quinolizidine enaminone 27. | |
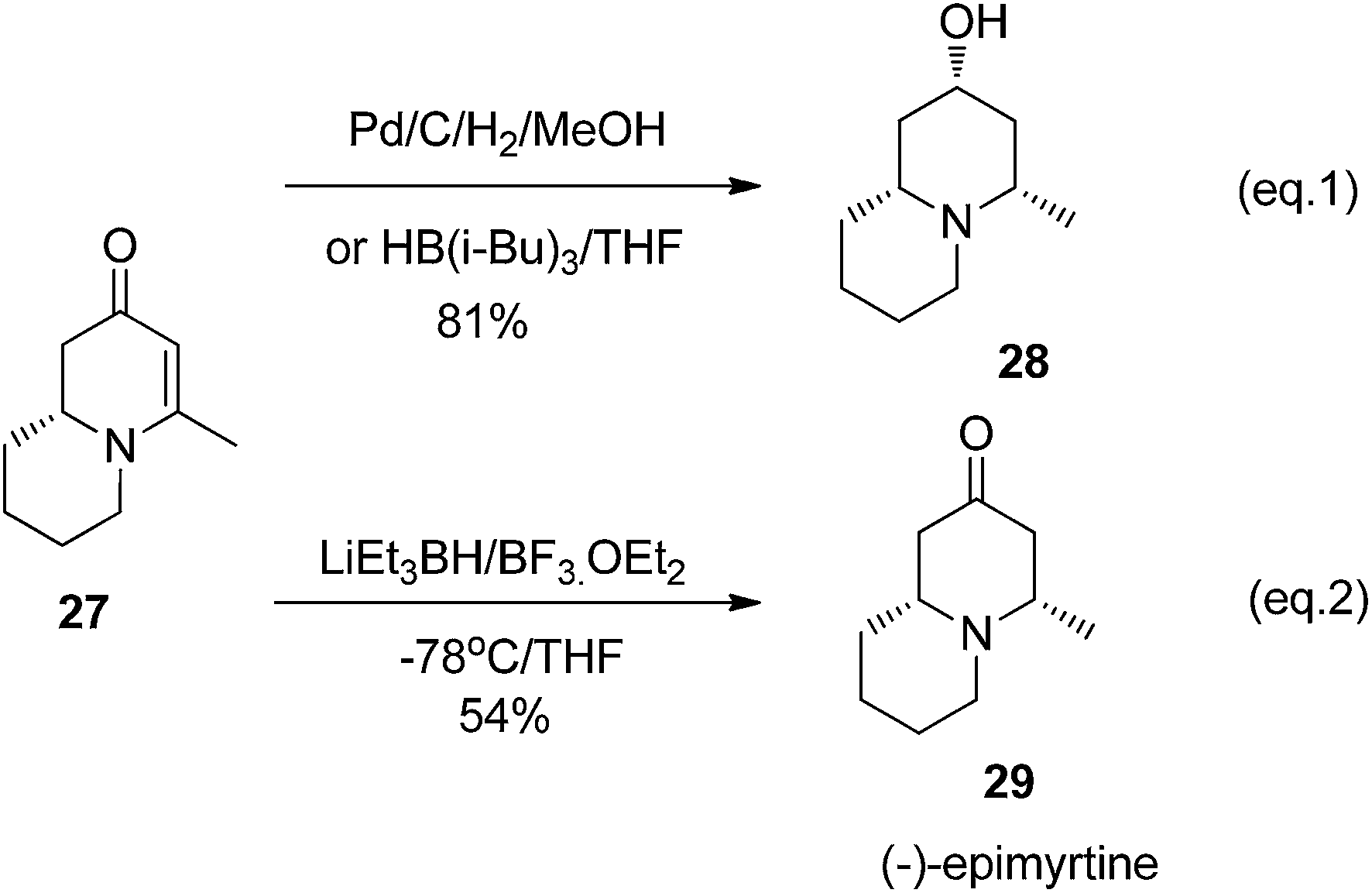
The reduction of cyclic enanminone 27 was first explored. It was found that under the conditions of either palladium-catalyzed hydrogenation in methanol or treating L-selectride (LiBui3BH) in THF, both alkene and carbonyl were reduced affording cis-2-hydroxyl-4-methly quinolizidine 28 as product (Scheme 6, eqn (1)). Similarly as in the reduction of 2,3-dihydropyridinone, the reduction on quinolizidine enanminone also proceeded in stereoselective manner which is in agree with literature precedents.26 When the reduction was carried out with “super hydride” (LiEt3BH) in presence of BF3·Et2O in THF, the alkene functionality was selectively reduced, giving 29 as natural quinolizidine alkaloid (−)-epimyrtine as the product in good yield27 (Scheme 6, eqn (2)). The results provided a concise approach for the enantioselective synthesis of such natural alkaloid.28
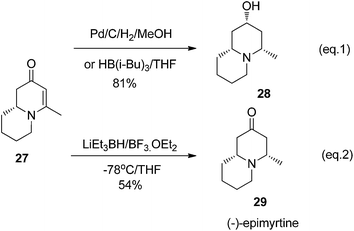 |
| | Scheme 6 Reduction of quinolizidine enaminone 27. | |
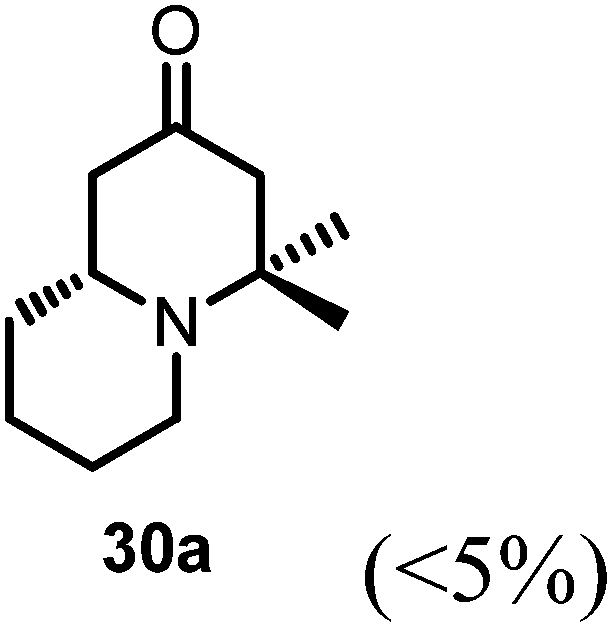
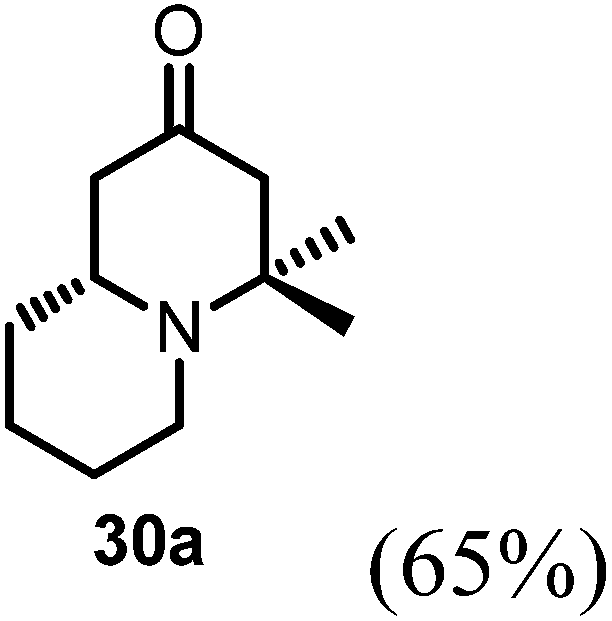
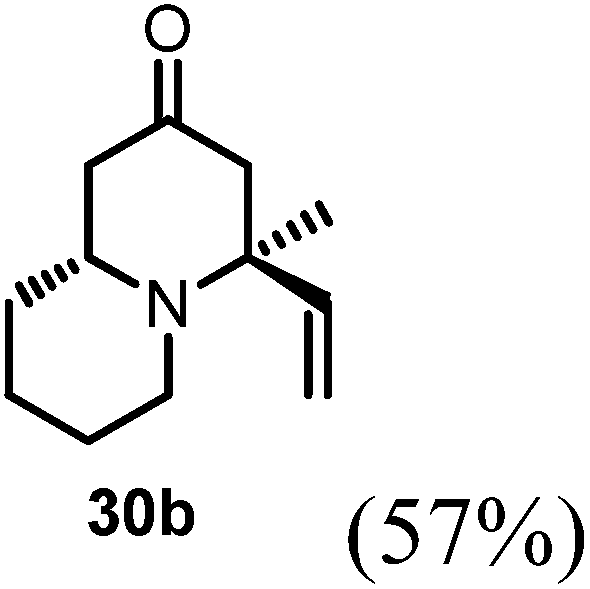
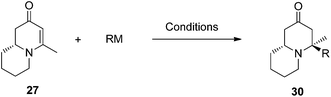
Conjugate additions to quinolizidine enaminone 27 were also explored. Although direct conjugate addition of Grignard reagents to cyclic enaminones has been previously reported,29 in our hands, treatment of 27 with methyl magnesium bromide did not yield expected product (Table 1, entry 1). Similarly, when methyl cuprate was employed, only a trace amount of product 30a was observed (Table 1, entry 2). However, when methyl Grignard addition was carried out in the presence of TMS-Cl, 1,4-conjugate addition went smoothly giving adduct 30 in good yield30 (Table 2, entry 3). Under similar conditions, conjugate additions by vinyl and allyl Grignard reagents were also performed (Table 2, entries 4 & 5). As the similar examples reported in literature, such conjugate addition on quinolizidine enaminone proceeded in stereoselective manner by generating a quaternary chiral carbon in the product (Scheme 7).31
Table 2 Conjugate addition reactions of quinolizidine enaminone 27
| Entry |
RM |
Conditions |
Products (yield) |
| 1 |
MeMgBr |
THF (0-RT) |
No reaction |
| 2 |
MeMgBr/CuI (Me2CuLi) |
THF (0-RT) |
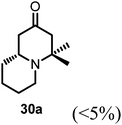
|
| 3 |
MeMgBr |
THF/TMS-Cl (3 eq.) (0-RT) |
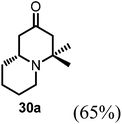
|
| 4 |
VinylMgBr |
THF/TMS-Cl (3 eq.) (0-RT) |
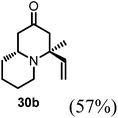
|
| 5 |
AllylMgBr |
THF/TMS-Cl (3 eq.) (0-RT) |
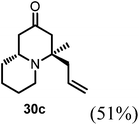
|
 |
| | Scheme 7 | |
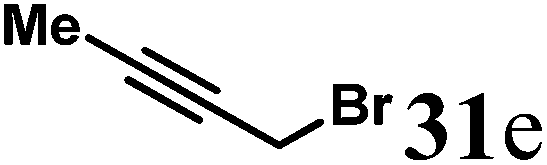
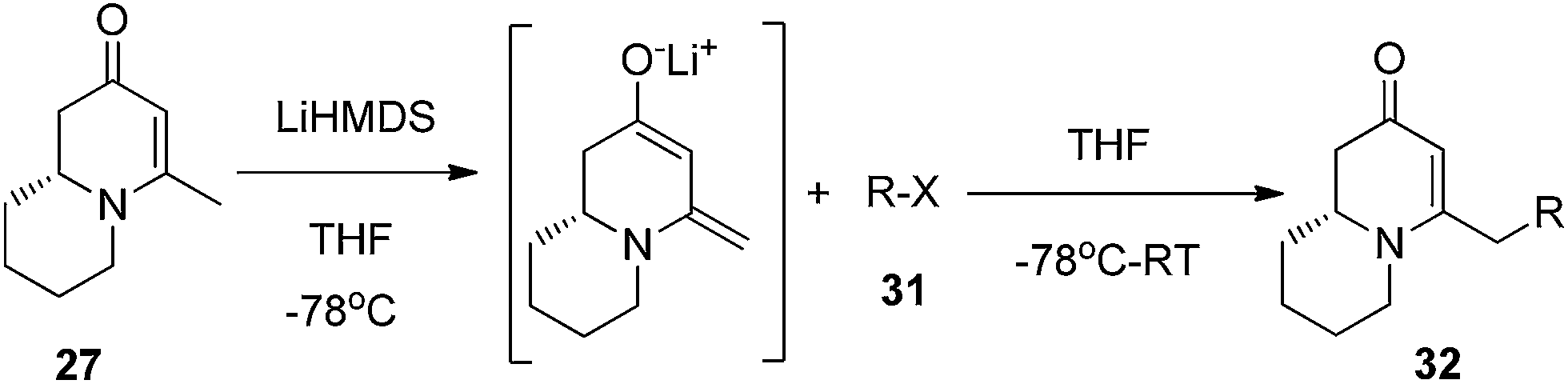
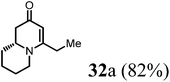
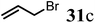
Finally, to further probe structural diversification, alkylation reaction on the methyl side chain of 27 was investigated. It was found that a corresponding enolate can be generated by treating 27 with LiN(SiMe3)2 (LiHMDS) in THF at low temperature. By subsequently treating such enolate with alkylating agents 31, corresponding alkylation products 32 could be obtained smoothly. The results of such reaction were summarized in Table 3. The reactions gave moderate to good yields by showing the tolerance toward different functional groups. No epimerization was observed in such enolate alkylation. Such alkylation reaction led to the side chain extension and provided opportunities to synthesize more structural diversified quinolizidine-based compound beyond α methyl substituted type (Scheme 8).
Table 3 Alkylation of quinolizidine enaminone 27
| Entry |
RX |
32 (yield) |
| 1 |
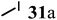
|
|
| 2 |
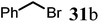
|
|
| 3 |
|
|
| 4 |
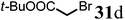
|
|
| 5 |
|
|
 |
| | Scheme 8 | |
Conclusion
In summary, inspired by the biosynthesis pathway of natural piperidine-based alkaloids, a general and practical approach to synthesize multi-substituted chiral piperidine was developed via a stereoselective three-component vinylogous Mannich-type reaction (VMR) by using 1,3-bis-trimethylsily enol ether 7 as a dienolate. The corresponding VMR adduct was chiral 2,3-dihydropyridinones 9 which played the role of cornerstone in building new targeted chiral piperidine compounds. The efficiency of such stereoselective synthesis approach was exampled in developing novel synthesis of bioactive natural alkaloids: dendrobate alkaloids (+)-241D; (−)-241D, and isosolenopsin A in highly concise manners. Beyond simple piperidine compound synthesis, the method also provided rapid route for chiral quinolizidine construction. When pre-functionalized substrate aldehyde 24 was employed, the corresponding VMR adduct could cyclize to give versatile quinolizidine cyclic enaminone 27. The different types transformations carried out on such polyfunctional intermediate gave rise a variety of new chiral quinolizidine compounds, including natural alkaloid (−)-epimyrtine. We believe the presented VMR approach offers a general; stereoselective and efficient way to assemble multi-substituted chiral piperidine-based compound in organic synthesis.
Experimental section
General methods
All commercial reagents and solvents were used without purification. 1H and 13C NMR spectra were recorded on a Bruker 400 MHz spectrometer using TMS as the internal standard (0 ppm). TLC analyses were carried out on aluminum sheets precoated with silica gel 60 F254, and UV radiation was used for detection. Flash column chromatography was performed on silica gel (SiliaFlash F60, 230–400 mesh). LC/MS analysis was performed on an Agilent 1100 series system equipped with an Agilent 1100 series binary pump, Agilent 1100 series autosampler, Agilent 1100 series DAD UV detector, Agilent 1100 series single quadrupole mass spectrometer with ESI source, and a SEDEX 75 ELSD. The mass spectrometer was set to scan from 100 to 1000 AMU. Mass spectrometric data were acquired in the positive ionization mode. The mobile-phase solvents used were (A) 0.05% aq. TFA; and (B) 0.035% TFA in MeCN. The total mobile phase flow rate was 1.0 mL min−1. The gradient was 10–90% in 3 min with an isocratic hold of 100% mobile-phase B for 0.49 min at the end of the gradient. A Waters Atlantis T3 (5 μm, 2.1 × 50 mm) column was used. Chemical shifts of NMR spectra are reported in ppm, multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). All vinylogous Mannich reactions were carried out in oven-dried glassware under air atmosphere. 1,3-Bis-trimethylsily enol ether 7 was prepared freshly following the procedure from literature.10d Diastereo-selectivities of VMR described in the manuscript were determined both by HPLC and NMR.
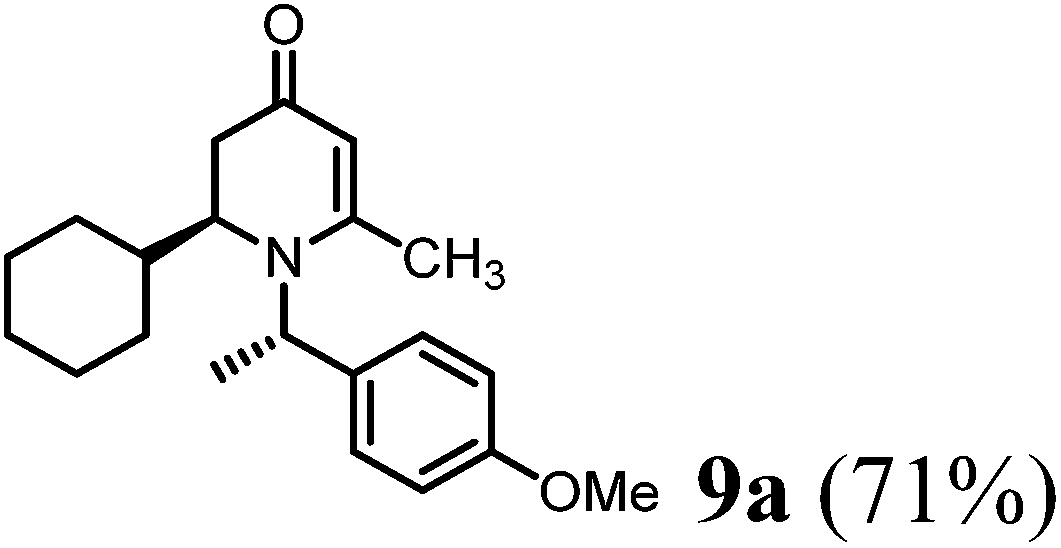
(S)-2-Cyclohexyl-1-((S)-1-(4-methoxyphenyl)ethyl)-6-methyl-2,3-dihydropyridin-4(1H)-one (9a).
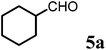
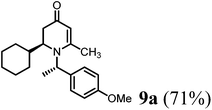
To a round-bottom flask contains (R)-1-(4-methoxyphenyl)ethanamine (6b) (151 mg, 1 mmol, 1 eq.) in dried DCM (0.1 M) solution added 4-AMS (500 mg mmol−1) followed by cyclohexane-carbaldehyde (5a) (112 mg, 1 mmol, 1 eq.). After stirring at room temperature for 10 min, 1,3-bis-trimethylsily enol ether 3 (292 mg, 1.2 mmol, 1.5 eq.) was added and the mixture solution was cooled to −78 °C. Tin(II) triflate (412 mg, 1 mmol, 1 eq.) was then added and the reaction was stirred at this temperature for 8 h. The reaction temperature was raised to 0 °C and kept the same temperature overnight. LC-MS analysis showed the mixture of 8a and 9a as new products (∼2/1). The reaction was quenched with saturated aqueous solution of sodium bicarbonate and removed solid via filtration. The reaction mixture was then extracted with DCM (15 mL × 5). The combined organic phase was treated with acetic acid (0.1 mL) and the resulting solution was stirred at room temperature for 1 h until 8a disappeared from LC-MS analysis. The solution was then basified by treating with saturated aqueous solution of sodium bicarbonate and washed with brine and dried over sodium sulfate. After concentration, the crude product was purified by ISCO silica gel chromatography (ethyl acetate/hexane) to afford the product as colorless oil (232 mg, 71% yield). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 0.87 (m, 1H); 1.30 (m, 4H); 1.41 (m, 1H); 1.55 (d, 3H, J = 6.8 Hz, CH3–); 1.70 (m, 5H); 2.02 (m, 2H); 2.07 (s, 3H, CH3–); 2.97 (m, 1H); 3.70 (s, 3H, OCH3); 4.87 (s, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ); 4.94 (q, 1H, J = 6.8 Hz); 6.80 (m, 2H); 7.13 (d, 2H, J = 8.4 Hz). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 18.8; 21.8; 26.2; 26.5; 26.6; 29.7; 30.6; 36.6; 40.9; 55.3; 558; 57.3; 102.5; 114.1; 127.5; 133.8; 159.1; 161.6; 191.9. LC-MS: 100% (purity), m/z: 328 (M + 1). Calcd for C21H30NO2 (M + H): 328.22765; found: 328.2272.
); 4.94 (q, 1H, J = 6.8 Hz); 6.80 (m, 2H); 7.13 (d, 2H, J = 8.4 Hz). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 18.8; 21.8; 26.2; 26.5; 26.6; 29.7; 30.6; 36.6; 40.9; 55.3; 558; 57.3; 102.5; 114.1; 127.5; 133.8; 159.1; 161.6; 191.9. LC-MS: 100% (purity), m/z: 328 (M + 1). Calcd for C21H30NO2 (M + H): 328.22765; found: 328.2272.
Similar procedure was applied to synthesize compound 9a–h; 14; 15; 17.
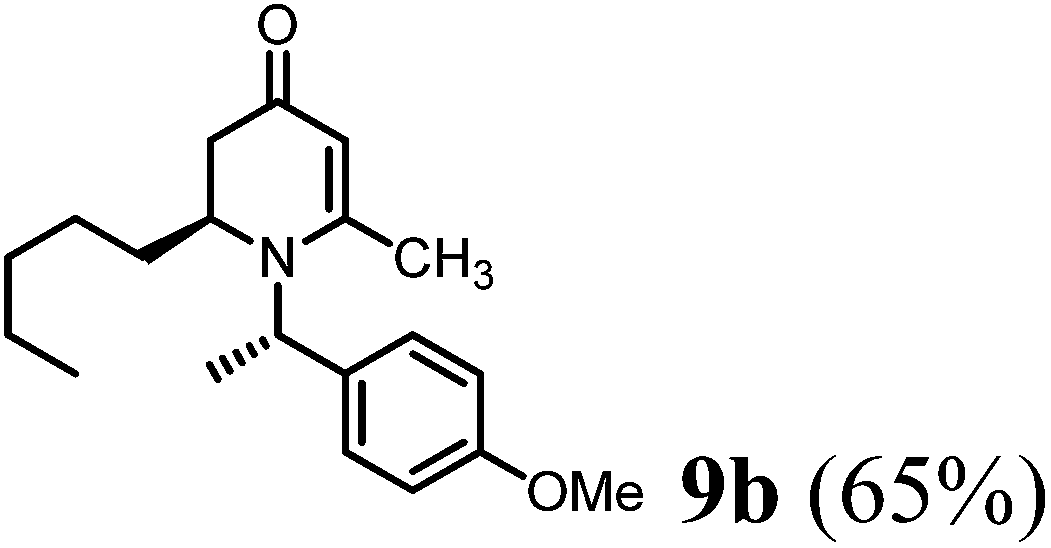
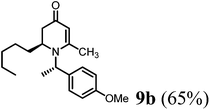
(R)-1-((S)-1-(4-Methoxyphenyl)ethyl)-6-methyl-2-pentyl-2,3-dihydropyridin-4(1H)-one (9b).
Colorless oil (yield, 65%). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 0.78 (t, 3H, J = 6.8 Hz, CH3); 1.05 (m, 1H); 1.18 (m, 5H); 1.25 (m, 2H); 1.56 (d, 3H, J = 6.8 Hz, CH3–); 2.01 (d, 1H, J = 16.58 Hz); 2.20 (dd, 1H, J1 = 6 Hz, J2 = 16.51 Hz); 2.07 (s, 3H, CH3–); 3.16 (m, 1H); 3.75 (s, 3H, OCH3); 4.87 (s, 1H, CH = ); 4.98 (q, 1H, J = 6.8 Hz); 6.83 (m, 2H); 7.16 (d, 2H, J = 8.4 Hz). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 14.0; 17.5; 21.5; 22.6; 25.7; 30.1; 31.7; 38.0; 52.9; 55.3; 55.4; 99.9; 100.0; 114.1; 127.6; 133.4; 159.1; 159.9; 190.6. LC-MS: 100% (purity), m/z: 316 (M + 1). Calcd for C20H30NO (M + H): 316.2276; found: 316.2271.
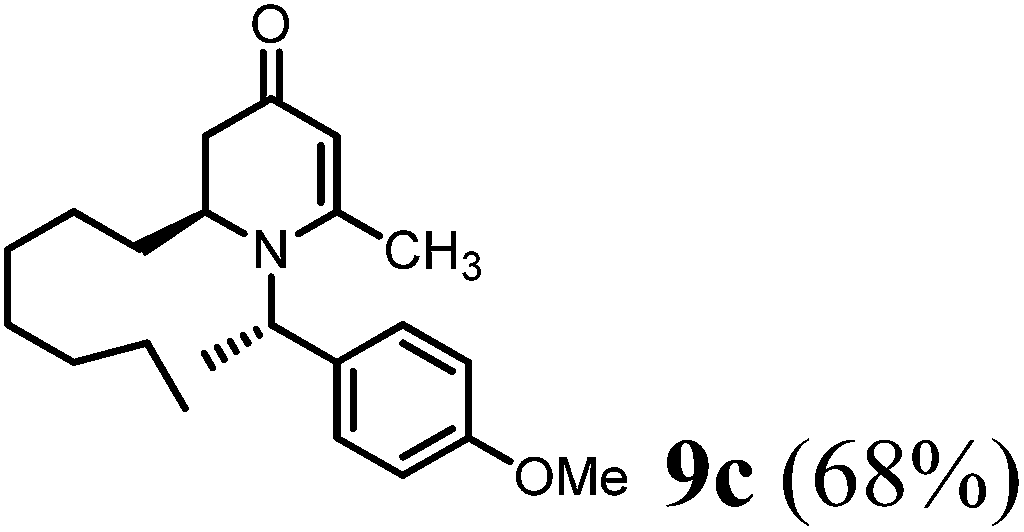
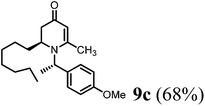
(R)-2-Heptyl-1-((S)-1-(4-methoxyphenyl)ethyl)-6-methyl-2,3-dihydropyridin-4(1H)-one (9c).
Colorless oil (yield, 68%). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 0.79 (t, 3H, J = 6.88 Hz, CH3); 1.16 (m, 12H); 1.56 (d, 3H, J = 7.06 Hz, CH3–); 1.98 (d, 1H, J = 16.52 Hz); 2.19 (dd, 1H, J1 = 5.94 Hz, J2 = 16.49 Hz); 2.08 (s, 3H, CH3–); 3.16 (m, 1H); 3.74 (s, 3H, OCH3); 4.86 (s, 1H, CH); 4.97 (q, 1H, J = 6.8 Hz); 6.83 (m, 2H); 7.16 (d, 2H, J = 8.69 Hz). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 14.1; 17.5; 21.5; 22.6; 25.9; 29.2; 29.5; 30.1; 31.7; 38.0; 52.9; 55.3; 55.4; 99.9; 114.1; 127.7; 133.3; 159.1; 159.9; 190.6. LC-MS: 100% (purity), m/z: 344 (M + 1). Calcd for C22H34NO2 (M + H): 344.2589; found: 344.2584.
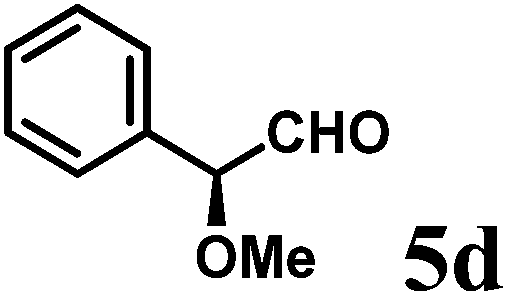
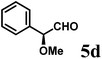
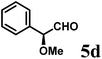
(S)-2-((S)-Methoxy(phenyl)methyl)-1-((S)-1-(4-methoxyphenyl)ethyl)-6-methyl-2,3-dihydropyridin-4(1H)-one (9d-I).
Colorless oil (yield, 59%). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 0.45 (d, J = 7.14 Hz, 3H); 1.33 (dd, J = 5.53 Hz, J2 = 16.92 Hz, 1H); 2.04 (s, 3H); 2.41 (d, J = 16.89 Hz, 1H); 3.11 (s, 3H); 3.26 (ddd, J1 = 1.62 Hz; J2 = 5.52 Hz; J3 = 8.80 Hz, 1H); 3.70 (s, 3H, OCH3); 4.50 (d, J = 8.80 Hz, 1H); 4.54 (q, J = 7.14 Hz, 1H); 5.09 (s, 1H); 6.74 (m, 2H); 6.98 (m, 2H); 7.18 (dd, J1 = 7.25, J2 = 8.83 Hz; 2H); 7.26 (m, 1H); 7.30 (m, 2H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 16.1; 21.7; 36.9; 55.3; 56.1; 57.8; 79.2; 104.4; 114.0; 127.5; 127.6; 128.1; 128.3; 133.5; 140.2; 159.1; 161.5; 192.6. LC-MS: 100% (purity), m/z: 366 (M + 1). Calcd for C23H28NO3 (M + H): 366.2069; found: 366.2065.
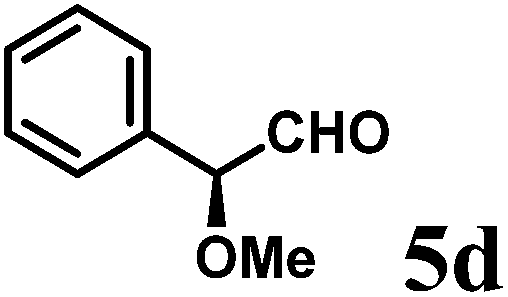
(R)-2-((S)-Methoxy(phenyl)methyl)-1-((R)-1-(4-methoxyphenyl)ethyl)-6-methyl-2,3-dihydropyridin-4(1H)-one (9d-II).
Colorless oil (yield, 64%). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 1.47 (dd, J1 = 12.13 Hz; J2 = 29.37 Hz, 1H); 1.69 (d, J = 7.21 Hz, 3H); 1.97 (m, 1H); 2.08 (s, 3H); 3.08 (s, 3H); 3.44 (dd, J1 = 6.88 Hz; J2 = 8.23 Hz, 1H); 3.71 (s, 3H); 4.49 (d, J = 9.19 Hz, 1H); 4.95 (s, 1H); 5.01 (q, J = 7.21 Hz, 1H); 6.78 (m, 2H); 7.20 (m, 7H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 19.3; 21.7; 36.5; 55.3; 56.9; 57.1; 58.4; 79.6; 102.0; 114.1; 127.6; 127.8; 128.2; 128.6; 134.3; 139.1; 159.0; 161.0; 190.5. LC-MS: 100% (purity), m/z: 366 (M + 1). Calcd for C23H28NO3 (M + H): 366.20692; found: 366.2065.
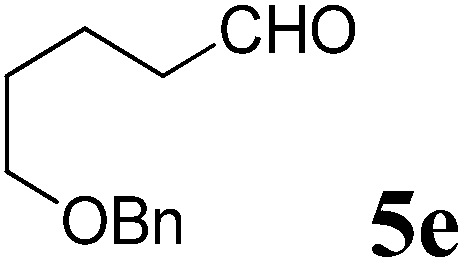
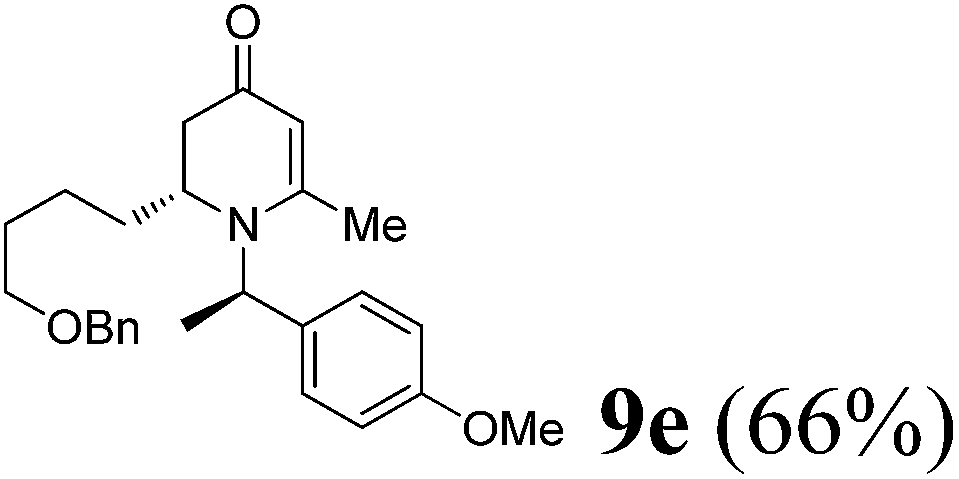
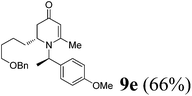
(R)-2-(4-(Benzyloxy)butyl)-1-((R)-1-(4-methoxyphenyl)ethyl)-6-methyl-2,3-dihydropyridin-4(1H)-one (9e).
Colorless oil (yield, 66%). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 1.19–1.41 (m, 4H); 1.48 (m, 2H); 1.52 (d, J = 7.04 Hz, 3H); 1.95 (d, J = 5.09 Hz, 1H); 1.99 (m, 1H); 2.08 (s, 3H); 2.18 (dd, J1 = 5.94 Hz; J2 = 16.54 Hz); 3.17 (m, 1H); 3.34 (m, 2H); 3.71 (s, 3H, OCH3); 4.39 (s, 2H); 4.86 (s, 1H); 4.95 (q, J = 7.04 Hz, 1H); 6.81 (m, 2H); 7.13 (m, 2H); 7.23 (m, 5H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 14.2; 17.5; 21.5; 22.7; 29.6; 29.8; 37.9; 52.8; 55.3; 55.5; 70.1; 73.0; 100.0; 114.1; 127.5; 127.64; 127.67; 127.70; 127.74; 128.4; 133.3; 138.5; 159.1; 159.9; 190.5. LC-MS: 100% (purity), m/z: 408 (M + 1). Calcd for C26H34NO3 (M + H): 408.2538; found: 408.2533.
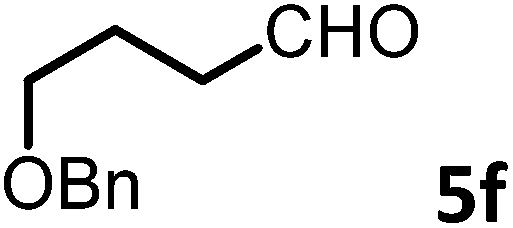
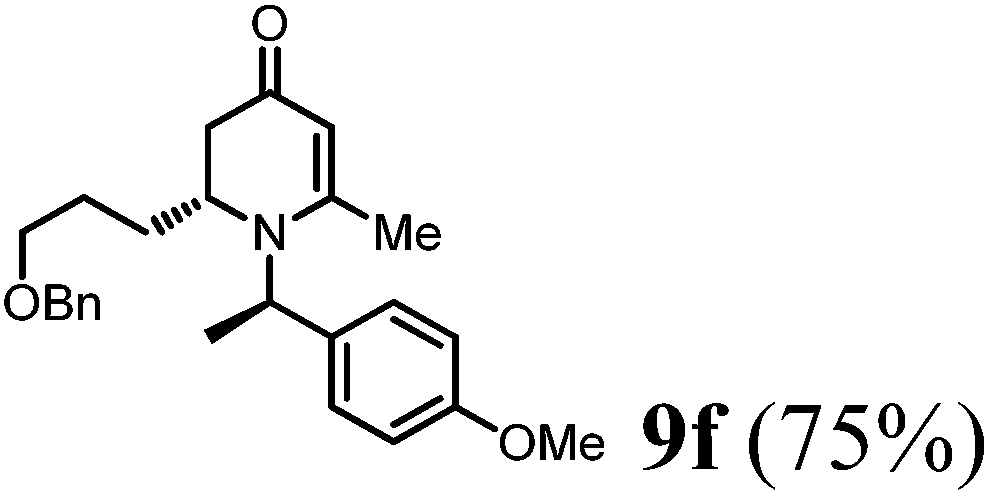
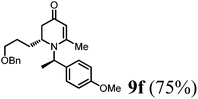
(R)-2-(3-(Benzyloxy)propyl)-1-((R)-1-(4-methoxyphenyl)ethyl)-6-methyl-2,3-dihydropyridin-4(1H)-one (9f).
Colorless oil (yield, 75%). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 1.39–1.24 (m, 1H); 1.47–1.40 (m, 1H); 1.55 (d, J = 7.20 Hz, 3H); 1.98–1.94 (m, 2H); 2.09 (s, 3H); 2.26–2.13 (m, 1H); 3.29–3.08 (m, 1H); 3.50–3.29 (m, 2H); 3.72 (s, 3H, OCH3); 4.38 (s, 2H); 4.87 (s, 1H); 4.94 (q, J = 7.20 Hz, 1H); 6.79 (m, 2H); 7.10 (m, 2H); 7.21 (m, 5H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 190.4; 160.0; 159.1; 138.4; 133.3; 128.4; 127.7; 127.6; 127.5; 114.1; 100.0; 72.9; 69.8; 55.5; 55.3; 52.5 38.0; 26.7; 25.9; 21.5; 17.4. LC-MS: 100% (purity), m/z: 394 (M + H). Calcd for C25H32NO3 (M + H): 394.2382 found: 394.2377.
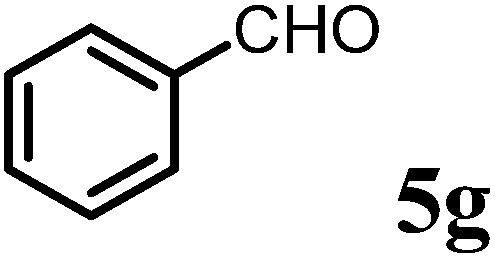
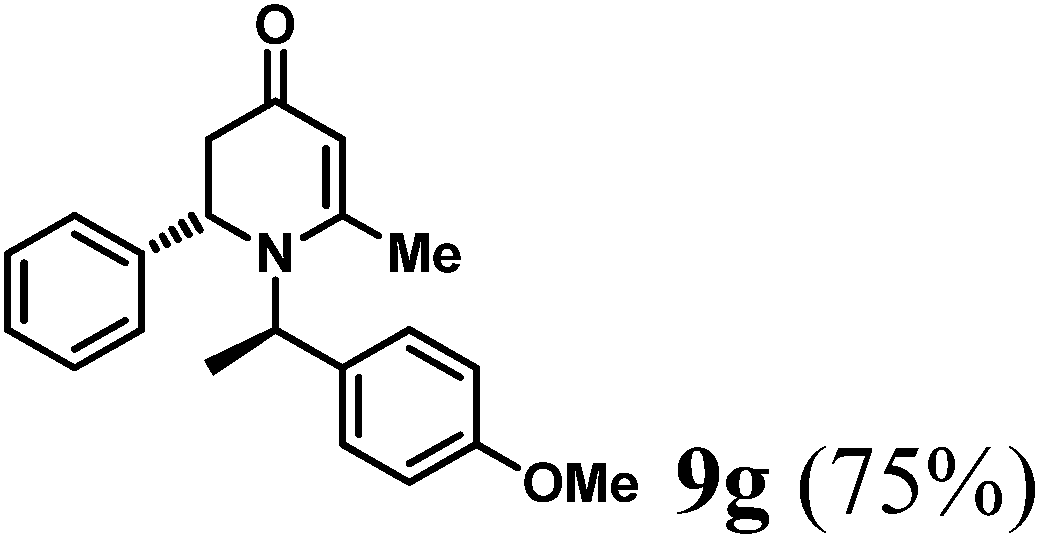
(R)-1-((R)-1-(4-Methoxyphenyl)ethyl)-6-methyl-2-phenyl-2,3-dihydropyridin-4(1H)-one (9g).
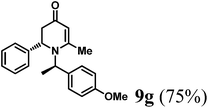
Colorless oil (yield, 75%). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 1.24 (d, J = 7.2 Hz, 3H), 2.18 (d, J = 16.3 Hz, 1H); 2.23 (s, 3H); 2.71 (dd, J1 = 7.53 Hz; J2 = 16.35 Hz, 1H); 3.739 (s, 3H); 4.42 (m, 1H); 4.96 (s, 1H); 5.12 (q, J = 6.97 Hz, 1H); 6.85 (m, 2H); 7.11 (m, 3H); 7.18 (m, 4H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 17.7; 21.6; 42.6; 55.4; 56.0; 56.1; 101.5; 114.2; 126.0; 127.3; 127.6; 128.6; 133.3; 140.6; 159.2; 161.6; 188.9. LC-MS: 100% (purity), m/z: 322 (M + 1). Calcd for C21H24NO2 (M + H): 322.1807; found: 322.1802.
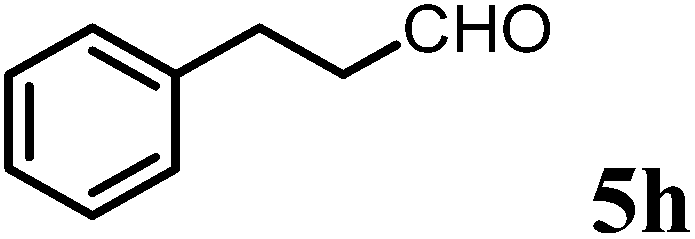
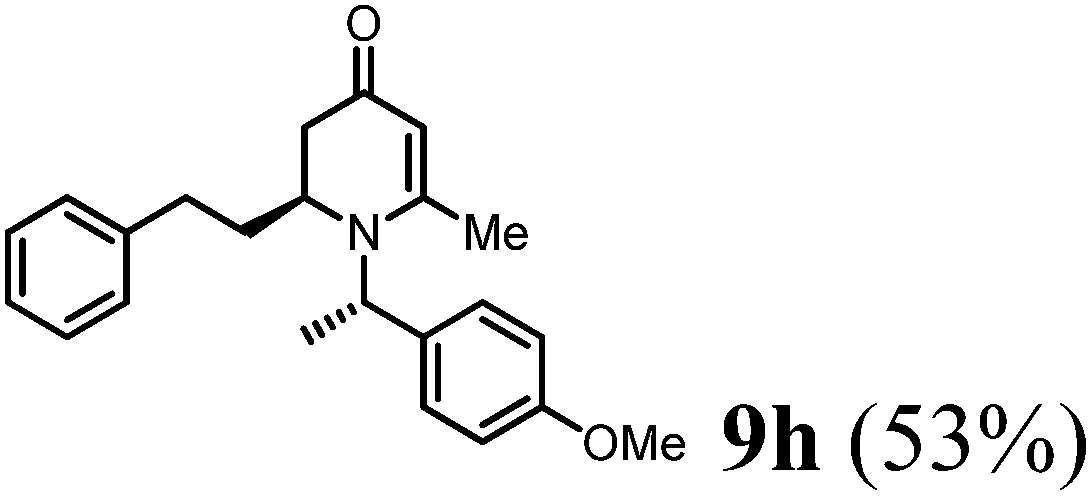
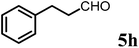
(S)-1-((S)-1-(4-Methoxyphenyl)ethyl)-6-methyl-2-phenethyl-2,3-dihydropyridin-4(1H)-one (9h).
Colorless oil (yield, 53%). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 7.19 (m, 2H); 7.13 (m, 1H); 7.04 (d, J = 7.16 Hz, 2H); 6.98 (d, J = 8.65 Hz, 2H); 6.76 (d, J = 8.71 Hz, 2H); 4.90 (q, J = 7.01 Hz, 1H); 4.87 (s, 1H); 3.73 (s, 1H, OCH3); 3.19 (m, 1H); 2.61 (m, 1H); 2.44 (m, 1H); 2.30 (m, 1H); 2.19 (m, 1H); 2.08 (s, 3H); 1.58 (m, 1H); 1.38 (d, J = 7.07 Hz, 3H, –CH3). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 190.3; 160.0; 159.1; 140.9; 132.9; 128.5; 127.7; 126.1; 114.0; 99.9; 55.6; 55.3; 51.5; 37.7; 31.9; 31.7; 21.5; 17.2. LC-MS: 100% (purity), m/z: 335 (M + 1). Calcd for C23H28NO2 (M + H): 350.2120; found: 350.2115.
(S)-1-((S)-1-(4-Methoxyphenyl)ethyl)-6-methyl-2-nonyl-2,3-dihydropyridin-4(1H)-one (14).
Colorless oil (yield, 76%). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 7.16 (d, J = 8.57 Hz, 2H); 6.82 (d, J = 8.79 Hz, 2H); 4.97 (q, J = 6.98 Hz, 1H); 4.86 (s, 1H); 3.73 (s, 3H); 3.16 (m, 1H); 2.19 (dd, Ja = 5.74 Hz, Jb = 16.53 Hz 1H); 2.08 (s, 3H); 1.97 (m, 1H); 1.55 (d, J = 6.98 Hz, 3H); 1.31–1.03 (m, 14H); 0.79 (t, J = 6.90 Hz, 3H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 190.6; 159.9; 159.1; 133.3; 127.6; 114.0; 99.8; 55.4; 55.3; 52.8; 38.0; 31.8; 30.1; 29.6; 29.5; 29.2; 25.9; 22.6; 21.5; 17.5; 14.1. LC-MS: 100% (purity), m/z: 372 (M + 1). Calcd for C24H38NO2 (M + H): 372.2902; found: 372.2897.
(R)-1-((R)-1-(4-Methoxyphenyl)ethyl)-6-methyl-2-nonyl-2,3-dihydropyridin-4(1H)-one (15).
Colorless oil (yield, 69%). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 7.13 (d, J = 8.66 Hz, 2H); 6.80 (d, J = 8.78 Hz, 2H); 4.95 (q, J = 6.98 Hz, 1H); 4.84 (s, 1H); 3.71 (s, 3H); 3.14 (m, 1H); 2.17 (dd, Ja = 6.02 Hz, Jb = 16.62 Hz 1H); 2.06 (s, 3H); 1.95 (m, 1H); 1.53 (d, J = 6.98 Hz, 3H); 1.35–0.94 (m, 14H); 0.77 (t, J = 6.89 Hz, 3H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 190.6; 159.9; 159.1; 133.3; 127.6; 114.0; 99.8; 55.4; 55.3; 52.9; 38.0; 31.8; 30.1; 29.6; 26.5; 29.4; 29.2; 25.9; 22.6; 21.5; 17.5; 14.1. LC-MS: 100% (purity), m/z: 372 (M + 1). Calcd for C24H38NO2 (M + H): 372.2902; found: 372.2897.
(R)-1-((R)-1-(4-Methoxyphenyl)ethyl)-6-methyl-2-undecyl-2,3-dihydropyridin-4(1H)-one (17).
Colorless oil (yield, 66%). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 7.21 (d, J = 8.62 Hz, 2H); 6.88 (d, J = 8.62 Hz, 2H); 5.03 (q, J = 6.99 Hz, 1H); 4.92 (s, 1H); 3.97 (s, 3H, OMe); 3.22 (m, 1H); 2.25 (dd, J1 = 5.88 Hz, J2 = 16.49 Hz 1H); 2.14 (s, 3H); 2.04 (m, 2H); 1.61 (d, J = 6.99 Hz, 3H); 1.35–1.10 (m, 20H); 0.86 (t, J = 6.86 Hz, 3H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 190.6; 159.9; 159.1; 133.3; 127.6; 114.0; 99.9; 55.4; 55.3; 52.9; 38.0; 31.9; 30.1; 29.6; 29.5; 29.4; 29.3; 25.9; 22.7; 21.5; 17.5; 14.1. LC-MS: 100% (purity), m/z: 400 (M + 1). Calcd for C15H42NO2 (M + H): 400.32155; found: 400.3210.
(S)-6-Methyl-2-phenethyl-2,3-dihydropyridin-4(1H)-one (10).
To a flask contains (S)-1-((S)-1-(4-methoxyphenyl)ethyl)-6-methyl-2-phenethyl-2,3-dihydropyridin-4(1H)-one (9h) (75 mg, 0.21 mmol) added TFA (1.5 mL) and the solution was stirred at room temperature overnight. TFA was removed by vacuum and the residue was re-dissolved in DCM (10 mL). The solution was washed with saturated aqueous solution of sodium bicarbonate and brine and dried over sodium sulfate. After concentration, the crude product was purified by ISCO silica gel chromatography (DCM/methanol) to afford the product as colorless oil (46 mg, 100% yield). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 7.22 (m, 2H); 7.14 (m, 3H); 4.59 (s, 1H); 4.84 (s, 1H); 3.57 (sex, J = 6.18 Hz, 1H); 2.67 (m, 1H); 2.61 (m, 1H); 2.34 (m, 1H); 2.22 (dd, Ja = 12.5 Hz, Jb = 16.08 Hz, 1H); 1.93 (m, 1H); 1.85 (m, 1H); 1.81 (s, 3H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 192.3; 161.8; 140.8; 128.7; 128.3; 126.4; 99.1; 53.0; 41.1; 35.7; 31.9; 21.2. LC-MS: 100% (purity), m/z: 216 (M + 1). Calcd for C14H18NO (M + H): 216.1388; found: 216.1383.
(2R,4S,6S)-2-Methyl-6-phenethylpiperidin-4-ol (11).
To a flask contains methanol solution (0.1 M) of 1(S)-1-((S)-1-(4-methoxyphenyl)ethyl)-6-methyl-2-phenethyl-2,3-dihydropyridin-4(1H)-one (9h) (120 mg, 0.34 mmol) added powder of palladium on carbon (12 mg) cautiously under nitrogen stream. The flask was then washed and filled with hydrogen gas. The reaction was stirred at room temperature for 4 h under hydrogen atmosphere and checked by LC-MS until starting material disappeared. After filtration to remove catalyst, the solvent was removed by vacuum and the crude product was purified by ISCO silica gel chromatography (DCM/methanol) to afford the product as gray powder (35 mg, 77% yield). NMR (400 MHz, CD3OD, 25 °C): δ = 7.10 (m, 2H); 7.14 (m, 2H); 7.05 (t, J = 7.13 Hz, 1H); 3.46 (ddd, Ja = 4.54 Hz, Jb = 7.83 Hz, Jc = 11.08 Hz); 2.56 (m, 3H); 2.44 (m, 1H); 1.93 (m, 1H); 1.79 (m, 1H); 1.68 (m, 1H); 1.58 (m, 1H); 1.01 (d, J = 6.37 Hz, 3H, –Me); 0.91 (m, 2H). 13C-NMR (100 MHz, CD3OD, 25 °C): δ = 143.3; 129.5; 129.4; 126.9; 69.6; 55.6; 51.4; 43.9; 41.5; 39.3; 33.3; 22.1. LC-MS: 100% (purity), m/z: 220 (M + 1). Calcd for C14H22NO (M + H): 220.1701; found: 220.1696.
(2R,6R)-2-Methyl-6-phenethylpiperidine (12).
To Parr shaker reaction vessel contains (0.1 M) of 1(S)-1-((S)-1-(4-methoxyphenyl) ethyl)-6-methyl-2-phenethyl-2,3-dihydropyridin-4(1H)-one (5h) (120 mg, 0.34 mmol) in mixed solvent of methanol (5 mL) and acetic acid (5 mL) added powder of palladium on carbon (36 mg) cautiously under nitrogen stream. The reaction was then carried out by Parr shaker hydrogenation apparatus under 40 psi overnight. After filtration to remove catalyst, the solvent was removed by vacuum and the crude product was re-dissolved in mixture of chloroform and 2-propanol (3/1, 15 mL). The solution was washed with saturated aqueous solution of sodium bicarbonate and brine and dried over sodium sulfate. After removing the solvent, the crude product was purified by ISCO silica gel chromatography (DCM/methanol) to afford the product as colorless oil (yield, 61%) (29 mg, 43% yield). 1H-NMR (400 MHz, CD3OD, 25 °C): δ = 7.26–6.21 (m, 5H); 3.03 (m, 1H); 2.93 (m, 1H); 2.72–2.42 (m, 2H); 2.01 (m, 1H); 1.89 (m, 1H); 1.81 (m, 3H); 1.63 (m, 1H); 1.46 (m, 1H); 1.28 (m, 1H); 1.21 (d, J = 6.41 Hz, 3H, Me). 13C-NMR (100 MHz, CD3OD, 25 °C): δ = 141.9; 129.7; 129.4; 129.3; 127.4; 58.3; 54.8; 36.9; 32.3; 31.8; 29.2; 23.6; 19.8. LC-MS: 100% (purity), m/z: 204 (M + 1). Calcd for C14H22N (M + H): 204.1752; found: 204.1747.
(2S,4R,6R)-2-Methyl-6-nonylpiperidin-4-ol [(−)-241D].
Starting from 15 similar hydrogenation procedure as in preparing 11 was applied to give (−)-241D as gray solid (yield, 69%). 1H-NMR (400 MHz, CD3OD, 25 °C): δ = 3.66 (tt, J1 = 4.51 Hz, J2 = 11.09 Hz); 3.02 (m, 1H); 2.90 (m, 1H); 2.06 (m, 1H); 1.98 (m, 1H); 1.58 (m, 1H); 1.43 (m, 1H); 1.36–1.14 (m, 17H); 1.09 (dd, J1 = 12.35 Hz, J2 = 24.34 Hz, 2H); 0.80 (t, J = 6.85 Hz, 3H, Me). 13C-NMR (100 MHz, CD3OD, 25 °C): δ = 65.8; 55.2; 51.1; 40.1; 37.6; 33.7; 31.7; 29.2; 29.1; 29.0; 25.0; 22.4; 18.5; 13.1. LC-MS: 100% (purity), m/z: 242 (M + 1). Calcd for C15H32NO2 (M + H): 242.2483; found: 242.2478 [α]D = −5.5° (C, 0.62, MeOH).
(2R,4S,6S)-2-Methyl-6-nonylpiperidin-4-ol [(+)-241D].
Starting from 14 similar hydrogenation procedure as in preparing 11 was applied to give (+)-241D as gray solid (yield, 72%). 1H-NMR (400 MHz, CD3OD, 25 °C): δ = 3.72 (tt, J1 = 4.42 Hz, J2 = 11.06 Hz); 3.16 (m, 1H); 3.05 (m, 1H); 2.13 (m, 1H); 2.05 (m, 1H); 1.62 (m, 1H); 1.47 (m, 1H); 1.41–1.18 (m, 17H); 1.14 (m, 2H); 0.80 (t, J = 6.84 Hz, 3H, Me). 13C-NMR (100 MHz, CD3OD, 25 °C): δ = 66.5; 56.8; 52.9; 40.6; 38.1; 34.4; 33.1; 30.6; 30.52; 30.55; 30.4; 26.2; 23.8; 19.2; 14.5. LC-MS: 100% (purity), m/z: 242 (M + 1). Calcd for C15H32NO2 (M + H): 242.2483; found: 242.2478 [α]D = +5.8° (C, 0.69, MeOH).
(2S,6R)-2-Methyl-6-undecylpiperidine (isosolenopsin A).
Starting from 17 similar hydrogenation procedure as in preparing 12 was applied to give isosolenopsin A as colorless oil (yield, 45%). 1H-NMR (400 MHz, CD3OD, 25 °C): δ = 3.07 (m, 1H); 2.94 (m, 1H); 1.92 (m, 1H); 1.83 (m, 2H); 1.75–1.52 (m, 1H); 1.52–1.37 (m, 2H); 1.37–1.09 (m, 23H); 0.80 (t, J = 6.73 Hz, 3H, Me). 13C-NMR (100 MHz, CD3OD, 25 °C): δ = 57.4; 53.4; 33.5; 31.7; 30.3; 29.3; 29.2; 29.0; 27.7; 24.7; 22.3; 22.1; 18.1; 13.0. LC-MS: 100% (purity), m/z: 254 (M + 1). Calcd for C17H36N (M + H): 254.2847; found: 254.2842 [α]D = +9.7° (C, 0.71, CHCl3).
(R)-2-(4-Chlorobutyl)-1-((R)-1-(4-methoxyphenyl)ethyl)-6-methyl-2,3-dihydropyridin-4(1H)-one (25).
To a round-bottom flask containing (R)-1-(4-methoxyphenyl)ethanamine (6a) (151 mg, 1 mmol, 1 eq.) in dried DCM (0.1 M) solution was added 4-AMS (500 mg mmol−1) followed by 5-chloropentanal (24) (120 mg, 1 mmol, 1 eq.). After stirring at room temperature for 10 min, (1-methoxy-buta-1,3-dienyloxy)-trimethyl-silane (7) (292 mg, 1.2 mmol, 1.2 eq.) was added and the solution was cooled to −78 °C. Tin(II) triflate (412 mg, 1 mmol, 1 eq.) was then added and the reaction was stirred at this temperature for 8 h. The reaction temperature was raised to 0 °C and kept at the same temperature overnight. LC-MS analysis of the reaction showed the disappearance of the starting materials. The reaction was quenched with saturated aqueous solution of sodium bicarbonate and solids were removed via filtration. The reaction mixture was then extracted with DCM (5 × 15 mL). The combined organic phase was treated with acetic acid and the resulting solution was stirred at room temperature for 1 h until only compound 25 was observed from LC-MS analysis. The solution was then basified by treating with saturated aqueous solution of sodium bicarbonate, washed with brine and dried over sodium sulfate. After concentration, the crude product was purified by ISCO silica gel chromatography (ethyl acetate/hexane) to afford the product 25 (ethyl acetate/hexane = 5/1) as colorless oil (184 mg, 55% yield). 1HNMR (400 MHz, CDCl3, 25 °C): δ = 7.23 (d, J = 8.45 Hz, 2H); 6.90 (d, J = 8.80 Hz, 2H); 5.06 (q, J = 7.01 Hz, 1H); 4.95 (s, 1H); 3.82 (s, 3H); 3.50 (m, 2H); 3.25 (m, 1H); 2.28 (dd, Ja = 5.94 Hz, Jb = 16.58 Hz 1H); 2.17 (s, 3H); 2.04 (m, 1H); 1.72 (m, 2H) 1.63 (d, J = 7.07 Hz, 3H); 1.38 (m, 3H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 190.4; 160.0; 159.2; 133.1; 127.6; 114.1; 100.1; 55.4; 55.3; 52.7; 44.7; 38.0; 32.3; 29.4; 23.4; 21.5; 17.5. LC-MS: 100% (purity), m/z: 336 (M + 1). Calcd for C19H27ClNO2 (M + H): 336.1730; found: 336.1725.
(R)-2-(4-Chlorobutyl)-6-methyl-2,3-dihydropyridin-4(1H)-one (26).
To the flask containing compound 25 (120 mg, 0.36 mol) was added TFA (2 mL, 99%) at room temperature and the resulting solution was stirred at room temperature overnight. TFA was removed by vacuum and the residue was re-dissolved in DCM (10 mL). The solution was washed with saturated aqueous solution of sodium bicarbonate and brine and dried over sodium sulfate. After concentration, the crude product was purified by ISCO silica gel chromatography (DCM/methanol) to afford the product as colorless oil (72 mg, 100% yield). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 4.59 (s, 1H); 4.85 (s, 1H); 3.56 (m, 1H); 2.32 (dd, Ja = 5.07 Hz, Jb = 16.10 Hz); 2.19 (dd, Ja = 5.07 Hz, Jb = 16.10 Hz); 1.91 (s, 3H); 1.74 (m, 2H); 1.63 (m, 2H); 1.53 (m, 1H); 1.47 (m, 2H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 190.4; 162.5; 98.8; 53.0; 44.7; 40.9; 33.3; 32.2; 22.7; 21.2. LC-MS: 100% (purity), m/z: 202 (M + 1). Calcd for C10H17ClNO (M + H): 202.0998; found: 202.0993.
(R)-4-Methyl-7,8,9,9a-tetrahydro-1H-quinolizin-2(6H)-one (27).
To the DMF (5 mL) solution contained 26 (95 mg, 0.47 mmol) added sodium hydride (56 mg, 60%, 3 eq.) at 0 °C. The reaction was stirred at this temperature for 2 h before quenched with saturated aqueous solution of ammonium chloride. The mixture was extracted with ethyl acetate (10 mL × 3) and the combined organic phase was washed with brine and dried over sodium sulfate. After concentration, the crude product was purified by ISCO silica gel chromatography (ethyl acetate/hexane) to afford the product as colorless oil (74 mg, 96% yield). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 4.93 (s, 1H); 3.72 (m, 1H); 3.29 (m, 1H); 2.75 (td, J1 = 2.91 Hz, J2 = 12.76 Hz, 1H); 1.92 (s, 3H); 1.80 (m, 1H); 1.68 (m, 1H); 1.58 (m, 2H); 1.43 (m, 2H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 191.5; 163.0; 101.8; 58.6; 48.1; 42.9; 31.4; 25.8; 23.7; 21.2. LC-MS: 100% (purity), m/z: 166 (M + 1). Calcd for C10H16NO (M + H): 166.1231; found: 166.1226.
(2R,4S,9aR)-4-Methyloctahydro-1H-quinolizin-2-ol (28).
To flask contained 27 (85 mg, 0.51 mmol) in THF (3 mL) added L-selectride (1 M in THF, 2.5 mL, 5 eq.) at 0 °C. The reaction was stirred at this temperature for 2 h before quenching with saturated aqueous solution of sodium bicarbonate. The mixture was extracted with mixed solvent (chloroform/isopropanol 3/1, 10 mL × 3) and the combined organic phase was washed with brine and dried over sodium sulfate. After concentration, the crude product was purified by ISCO silica gel chromatography (chloroform/methanol) to afford the product as colorless oil (70 mg, 81% yield). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 4.13 (s, 1H, OH); 3.70 (m, 1H); 3.63 (m, 1H); 3.40 (m, 1H); 3.31 (m, 1H); 2.45 (m, 1H); 2.25 (t, J = 13.21 Hz, 1H); 2.15 (t, J = 12.77 Hz, 2H); 1.94 (m, 3H); 1.83 (m, 2H); 1.69 (m, 1H); 1.44 (m, 1H); 1.42 (d, J = 6.30 Hz, 3H, Me). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 61.8; 59.2; 56.1; 50.9; 38.6; 37.4; 30.1; 23.3; 22.6; 17.2. LC-MS: 100% (purity), m/z: 170 (M + 1). Calcd for C10H20NO (M + H): 170.1544; found: 170.1539.
(4S,9aR)-4-Methylhexahydro-1H-quinolizin-2(6H)-one (29).
To flask contained 27 (68 mg, 0.41 mmol) in THF (2 mL) added BF3·OEt2 (57 μL, 1.1 eq.) at −78 °C. The reaction was stirred at this temperature for 10 min before adding lithium triethylborohydride “super hydride” (1 M in THF, 0.49 mL, 1.2 eq.). After stirring at −78 °C for 2 h, the reaction was quenched with saturated aqueous solution of sodium bicarbonate. The mixture was extracted with mixed solvent (chloroform/isopropanol 3/1, 10 mL × 3) and the combined organic phase was washed with brine and dried over sodium sulfate. After concentration, the crude product was purified by ISCO silica gel chromatography (chloroform/methanol) to afford the product as colorless oil (37 mg, 54% yield). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 3.27 (m, 1H); 2.33 (m, 3H): 2.22 (m, 2H): 2.12 (t, J = 10.95 Hz, 1H); 1.78 (m, 1H); 1.68 (m, 2H); 1.58 (m, 2H); 1.36 (m, 1H); 1.20 (m, 1H); 1.14 (d, J = 5.72 Hz, 3H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 207.9; 61.6; 58.8; 50.5; 49.2; 48.1; 33.6; 25.4; 23.4; 20.2. LC-MS: 100% (purity), m/z: 168 (M + 1). Calcd for C10H18NO (M + H): 168.1388; found: 168.1383 [α]D = −16.9° (C, 0.28, CHCl3).
(R)-4,4-Dimethylhexahydro-1H-quinolizin-2(6H)-one (30a).
To the flask contained 27 (42 mg, 0.25 mmol) in dried THF (2 mL) added TMS-Cl (80 μL, 3 eq.) at 0 °C. The reaction was stirred at this temperature for 10 min before adding methyl magnesium bromide (2 M in ethyl ether, 0.18 mL, 1.5 eq.). After stirring at 0 °C for 2 h, the reaction temperature was allowed to raise to room temperature overnight. Quenched by saturated aqueous solution of sodium bicarbonate the reaction mixture was extracted with mixed solvent (chloroform/isopropanol 3/1, 10 mL × 3) and the combined organic phase was washed with brine and dried over sodium sulfate. After concentration, the crude product was purified by ISCO silica gel chromatography (chloroform/methanol) to afford the product as colorless oil (28 mg, 65% yield). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 3.0 (m, 1H); 2.48 (m, 2H); 2.15 (m, 2H); 1.99 (m, 2H); 1.60 (m, 3H); 1.45 (m, 1H); 1.27 (m, 1H); 1.10 (m, 1H); 1.13 (s, 3H); 0.81 (s, 3H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 209.1; 58.0; 55.8; 55.4; 48.2; 45.4; 34.6; 29.6; 25.8; 23.8; 15.8. LC-MS: 100% (purity), m/z: 182 (M + 1). Calcd for C11H20NO (M + H): 182.1544; found: 182.1539.
(4S,9aR)-4-Methyl-4-vinylhexahydro-1H-quinolizin-2(6H)-one (30b).
Starting from 27 similar addition procedure as in preparing 30a was applied to give 30b as colorless oil (yield, 57%). 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 5.82 (dd, J = 17.6, 11.1 Hz, 1H), 5.16 (d, J = 11.0 Hz, 1H), 5.01 (d, J = 17.5 Hz, 1H), 3.07 (m, 1H), 2.84–2.42 (m, 2H), 2.37 (m, 1H), 2.21 (m, 3H), 1.93–1.72 (m, 1H), 1.63 (m, 3H), 1.57–1.41 (m, 1H), 1.27 (s, 3H), 1.18 (m, 2H). 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 209.1; 137.1; 116.6; 61.2; 55.1; 51.9; 47.5; 44.9; 34.3; 26.5; 25.8; 23.3. LC-MS: 100% (purity), m/z: 194 (M + 1). Calcd for C12H19NO (M + H): 194.1544; found: 194.1540.
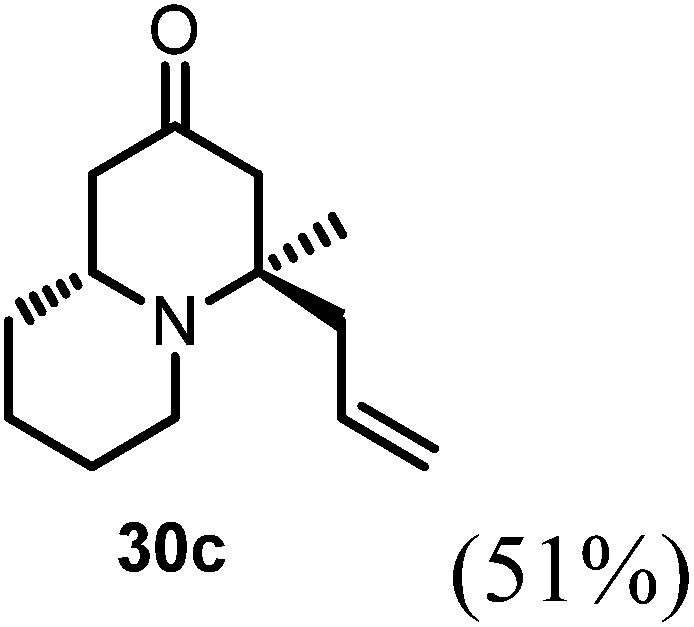
(4R,9aR)-4-Allyl-4-methylhexahydro-1H-quinolizin-2(6H)-one (30c).
Starting from 27 similar addition procedure as in preparing 30a was applied to give 14 as colorless oil (yield, 57%). 1H NMR (400 MHz, CDCl3, 25 °C) δ = 5.81–5.44 (m, 1H), 5.15–4.86 (m, 2H), 3.22–2.97 (m, 1H), 2.80–2.59 (m, 1H), 2.31 (s, 2H), 2.27–2.11 (m, 3H), 2.08–1.97 (m, 2H), 1.66 (d, J = 9.9 Hz, 2H), 1.31 (m, 2H), 1.18 (s, 2H), 1.15 (s, 3H). 13C NMR (101 MHz, CDCl3, 25 °C) δ = 209.33; 133.84; 118.66; 61.04; 55.72; 51.76; 48.41; 45.38; 35.28; 34.28; 26.91; 26.43; 24.13. LC-MS: 100% (purity), m/z: 208 (M + 1). Calcd for C13H22NO (M + H): 208.1701; found: 208.1696.
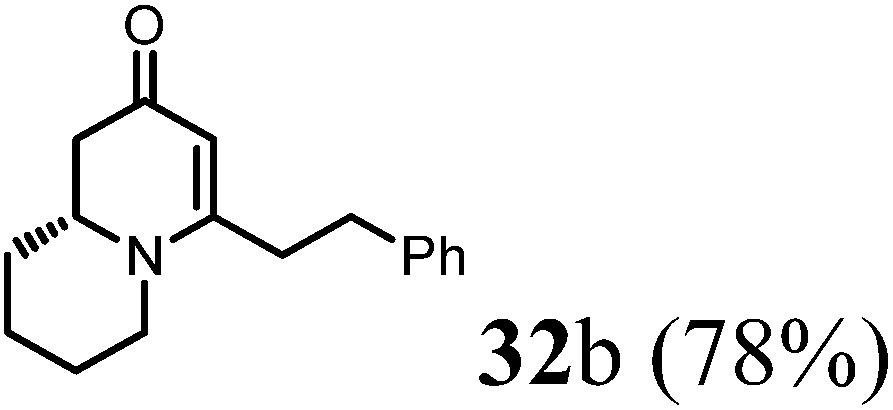
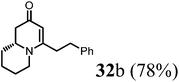
(1R,9aR)-1-Benzyl-4-methyl-7,8,9,9a-tetrahydro-1H-quinolizin-2(6H)-one (32b).
To the flask contained 27 (42 mg, 0.25 mmol) in dried THF (2 mL) added LiHMDS (0.3 mL, 1 M in THF) at −78 °C. The reaction was stirred at this temperature for 1 h before adding benzyl bromide (88 μL, 3 eq.). After stirring at 0 °C for 2 h, the reaction temperature was allowed to raise to room temperature overnight. Quenched by saturated aqueous solution of sodium bicarbonate the reaction mixture was extracted with mixed solvent (ethyl acetate, 10 mL × 3) and the combined organic phase was washed with brine and dried over sodium sulfate. After concentration, the crude product was purified by ISCO silica gel chromatography (hexane/ethyl acetate) to afford the product as colorless oil (49 mg, 78% yield).1H NMR (400 MHz, CDCl3, 25 °C) δ = 7.27–7.19 (m, 2H); 7.19–7.08 (m, 3H); 5.00 (s, 1H); 3.91–3.57 (m, 1H); 3.29 (dd, J = 5.9, 4.2 Hz, 1H); 2.97–2.62 (m, 3H); 2.62–2.38 (m, 3H); 2.23 (dd, J = 16.5, 10.7 Hz, 1H); 1.86–1.73 (m, 1H); 1.73–1.49 (m, 3H); 1.49–1.31 (m, 2H). 13C NMR (101 MHz, CDCl3, 25 °C) δ = 191.2; 165.2; 139.7; 128.1; 127.8; 126.1; 100.5; 76.9; 58.2; 47.6; 42.2; 35.1; 33.9; 31.0; 25.5; 23.3. LC-MS: 100% (purity), m/z: 256 (M + 1). Calcd for C17H22NO (M + H): 256.1701; found: 256.1696.
Starting from 27 similar alkylation procedure as in preparing 32b was applied to give 32a; 32c; 32d and 32e.
(R)-4-Ethyl-7,8,9,9a-tetrahydro-1H-quinolizin-2(6H)-one (32a).
Colorless oil (yield, 82%). 1H NMR (400 MHz, CDCl3, 25 °C) δ = 4.99 (s, 1H); 3.81–3.63 (m, 1H); 3.39–3.19 (m, 1H); 2.74 (td, J = 12.7, 2.9 Hz, 1H); 2.47 (dd, J = 16.6, 5.9 Hz, 1H); 2.29–2.18 (m, 2H); 2.17 (m, 1H); 1.79 (m 1H); 1.74–1.66 (m, 1H); 1.66–1.54 (m, 2H); 1.54–1.32 (m, 2H); 1.09 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3, 25 °C) δ = 191.2; 167.6; 99.4; 58.2; 47.3; 42.0; 31.0; 26.4; 25.5; 23.3; 11.9. LC-MS: 100% (purity), m/z: 180 (M + 1). Calcd for C11H18NO (M + H): 180.1388; found: 180.1383.
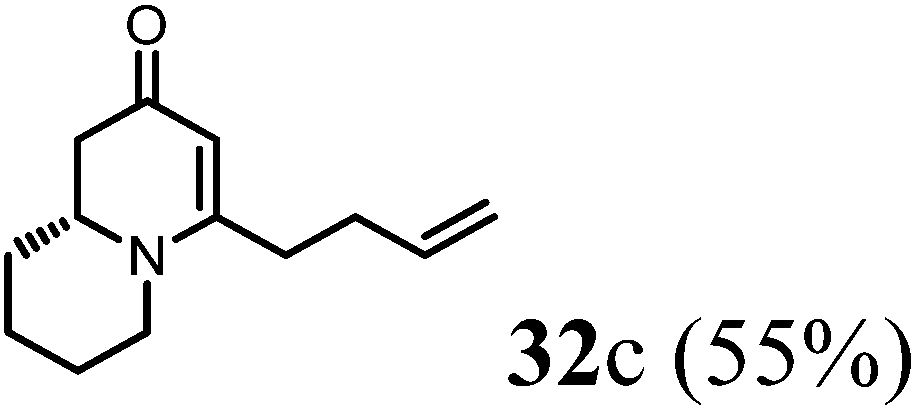
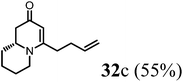
(R)-4-(But-3-en-1-yl)-7,8,9,9a-tetrahydro-1H-quinolizin-2(6H)-one (32c).
Colorless oil (yield, 55%). 1H NMR (400 MHz, CDCl3, 25 °C) δ = 5.96–5.72 (m, 1H); 5.21–4.93 (m, 3H); 3.94–3.64 (m, 1H); 3.49–3.18 (m, 1H); 2.82 (td, J = 12.7, 2.8 Hz, 1H); 2.53 (dd, J = 16.4, 5.8 Hz, 1H); 2.45–2.19 (m, 3H); 1.92–1.83 (m, 1H); 1.83–1.74 (m, 3H); 1.69 (dd, J = 6.8, 3.6 Hz, 2H); 1.61–1.41 (m, 2H). 13C NMR (101 MHz, CDCl3, 25 °C) δ = 191.8; 165.7; 136.5; 115.9; 101.1; 58.6; 48.1; 42.8; 33.2; 32.1; 31.5; 26.0; 23.8. LC-MS: 100% (purity), m/z: 205 (M + 1). Calcd for C13H20NO (M + H): 206.1545; found: 206.1540.
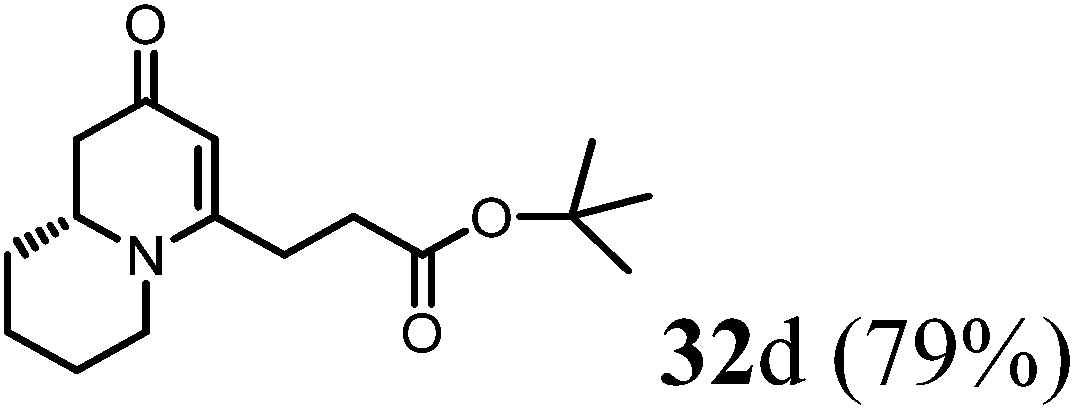
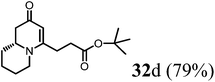
(R)-tert-Butyl-3-(2-oxo-2,6,7,8,9,9a-hexahydro-1H-quinolizin-4-yl)propanoate (32d).
Colorless oil (yield, 79%). 1H NMR (400 MHz, CDCl3, 25 °C) δ = 4.92 (s, 1H); 3.89–3.63 (m, 1H); 3.47–3.17 (m, 1H); 2.88–2.63 (m, 1H); 2.56–2.41 (m, 3H); 2.41–2.34 (m, 2H); 2.21 (dd, J = 16.4, 10.4 Hz, 1H); 1.86–1.75 (m, 1H); 1.75–1.65 (m, 1H); 1.65–1.52 (m, 2H); 1.52–1.39 (m, 2H); 1.38 (s, 9H, t-Bu). 13C NMR (101 MHz, CDCl3, 25 °C) δ = 190.7; 170.2; 163.5; 99.6; 80.2; 57.7; 47.0; 41.7; 32.4; 30.3; 27.6; 27.1; 27.1; 24.9; 22.7. LC-MS: 100% (purity), m/z: 280 (M + 1). Calcd for C16H26NO3 (M + H): 280.1913; found: 280.1908.
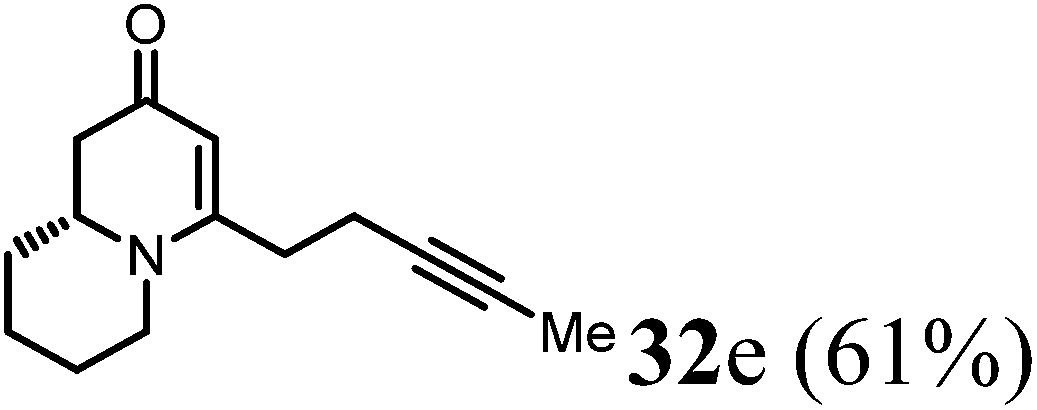
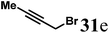
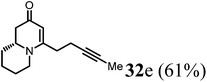
(R)-4-(Pent-3-yn-1-yl)-7,8,9,9a-tetrahydro-1H-quinolizin-2(6H)-one (32e).
Colorless oil (yield, 61%). 1H NMR (400 MHz, CDCl3, 25 °C) δ = 4.95 (s, 1H); 3.91–3.57 (m, 1H); 3.39–3.17 (m, 1H); 2.74 (td, J = 12.8, 2.8 Hz, 1H); 2.50–2.33 (m, 3H); 2.33–2.27 (m, 2H); 2.23 (dd, J = 16.5, 10.7 Hz, 1H); 1.89–1.74 (m, 1H); 1.71 (s, 3H); 1.69–1.65 (m, 1H); 1.65–1.51 (m, 2H); 1.51–1.33 (m, 2H). 13C NMR (101 MHz, 25 °C) δ = 191.9; 164.5; 101.0; 77.2; 76.9; 58.6; 48.1; 42.8; 33.2; 31.4; 25.9; 23.8; 17.9; 3.5. LC-MS: 100% (purity), m/z: 218 (M + 1). Calcd for C14H20NO (M + H): 218.1545; found: 218.1541.
Acknowledgements
Dedicated to ICCAS Emeritus Professor Dong Wang on the occasion of his 74th birthday. The author thanks Dr Perry Gordon and Mr Kevin Johnson for providing high resolution mass spectrometry data (HRMS) of all reported compounds. The author sincerely appreciates the management supports from Dr John Tellew; Dr Shifeng Pan; Dr Dean P. Phillips and Dr Valentina Molteni. The author is also grateful to Dr John Tellew and Dr Yahu A. Liu for helpful discussions and proof reading.
References
-
G. M. Struntz and J. A. Findlay, in The Alkaloids, ed. A. Brossi, Academic, New York, 1985, vol. 26, pp. 89–193 Search PubMed.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- For recent reviews see:
(a) S. Kallstrom and R. Leino, Bioorg. Med. Chem., 2008, 681 Search PubMed;
(b) M. G. P. Buffat, Tetrahedron, 2004, 1701 CrossRef CAS;
(c) F.-X. Felpin and J. Lebreton, Eur. J. Org. Chem., 2003, 3693 CrossRef CAS;
(d) S. Laschat and T. Dickner, Synthesis, 2000, 1781 CrossRef CAS.
- Y. Ying, H. Kim and J. Hong, Org. Lett., 2011, 13, 796–799 CrossRef CAS PubMed and references therein.
-
G. M. Strunz and J. A. Findlay, Pyridine and piperidine alkaloids, in The Alkaloids, ed. A. Brossi, Academic Press, New York, 1985, vol. 26, pp. 89–174 Search PubMed.
- For general reviews see:
(a)
P. M. Dewick, in Medicinal Natural Products, A Biosynthetic Approach, Wiley, 3rd revised edn, 2002 Search PubMed;
(b)
E. Szoke, E. Lemberkovics and L. Kursinszki, in Natural Products, Alkaloids Derived from Lysine: Piperidine Alkaloids, ed. K. G. Ramawat and J. M. Merillon, Springer, 2013, pp. 301–343 Search PubMed.
- Δ1-Piperideine is unstable and exists in solution as a complex diastereoisomeric mixture of a trimeric (major component) and monomeric form:
(a) A. Rouchaud and J. C. Braekman, Eur. J. Org. Chem., 2009, 2666 CrossRef CAS;
(b) C. Darwich, M. Elkhatib, G. Steinhauser and H. Delalu, Helv. Chim. Acta, 2009, 92, 98 CrossRef CAS;
(c) J. C. Guillemin, J. M. Denis, M. C. Lasne and J. L. Ripoll, Tetrahedron, 1988, 44, 4447–4455 CrossRef CAS.
- Biomimetic synthesis of piperidine-type alkaloids by using Δ1-piperideine was reported by Bella et al. see: M. R. Monaco, P. Renzi, D. M. Scarpino Schietroma and M. Bella, Org. Lett., 2011, 13, 4546–4549 CrossRef CAS PubMed.
-
(a) D. L. Comins and J. D. Brown, Tetrahedron Lett., 1986, 27, 4549 CrossRef CAS;
(b) D. L. Comins and L. A. Morgan, Tetrahedron Lett., 1991, 32, 5919 CrossRef CAS;
(c) D. L. Comins and H. Hong, J. Org. Chem., 1993, 58, 5035 CrossRef CAS;
(d) D. L. Comins and N. R. Benjelloun, Tetrahedron Lett., 1994, 35, 829 CrossRef CAS;
(e) D. L. Comins and Y. M. Zhang, J. Am. Chem. Soc., 1996, 118, 12248 CrossRef CAS;
(f) D. L. Comins, A. H. Libby, R. S. Al-awar and C. J. Foti, J. Org. Chem., 1999, 64, 2184 CrossRef CAS;
(g) D. L. Comins, M. J. Sandelier and T. A. Grillo, J. Org. Chem., 2001, 66, 6829 CrossRef CAS PubMed;
(h) R. Badorrey, C. Cativiela, M. D. Diaz de Villegas and J. A. Galvez, Tetrahedron, 2002, 58, 341 CrossRef CAS;
(i) N. S. Josephsohn, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, 4018 CrossRef CAS PubMed;
(j) D. L. Comins and J. J. Sahn, Org. Lett., 2005, 7, 5227 CrossRef CAS PubMed;
(k) R. T. Yu and T. Rovis, J. Am. Chem. Soc., 2006, 128, 12370 CrossRef CAS PubMed;
(l) B. J. Turunen and G. I. Georg, J. Am. Chem. Soc., 2006, 128, 8702 CrossRef CAS PubMed;
(m) C. A. Newman, J. C. Antilla, P. Chen, A. V. Predeus, L. Fielding and W. D. Wulff, J. Am. Chem. Soc., 2007, 129, 7216 CrossRef CAS PubMed;
(n) T. Andreassen, T. Haaland, L. K. Hansen and O. R. Gautun, Tetrahedron Lett., 2007, 48, 8413 CrossRef CAS;
(o) R. K. Friedman and T. Rovis, J. Am. Chem. Soc., 2009, 131, 10775 CrossRef PubMed;
(p) M. J. Niphakis, B. J. Turunen and G. I. Georg, J. Org. Chem., 2010, 75, 6793 CrossRef CAS PubMed;
(q) For a recent review: J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed.
- As a dianion synthons with 1,2-dielectrophiles
(a) P. Langer and V. Kohler, Chem. Commun., 2000, 1653–1654 RSC;
(b) E. Ulah, B. Appel, C. Fischer and P. Langer, Tetrahedron Lett., 2006, 62, 9694–9700 CrossRef;
(c) P. Langer and K. Valentin, Org. Lett., 2000, 2, 1597–1599 CrossRef CAS PubMed;
(d) E. Ullah, B. Appel, C. Fischer and P. Langer, Tetrahedron, 2006, 62, 9694–9700 CrossRef CAS;
(e) E. Holtz, V. Koehler, B. Appel and P. Langer, Eur. J. Org. Chem., 2005, 532–542 CrossRef CAS;
(f) I. Hussain, M. A. Yawer, M. Lau, T. Pundt, C. Fischer, H. Gorls and P. Langer, Eur. J. Org. Chem., 2008, 503–518 CrossRef CAS;
(g) G. Bose, E. Ullah and P. Langer, Chem.–Eur. J., 2004, 10, 6015–6028 CrossRef CAS PubMed;
(h) M. Lubbe, R. Klassen, T. Trabhardt, A. Villinger and P. Langer, Synlett, 2008, 15, 2331 Search PubMed;
(i) E. Bellur, H. Goerls and P. Langer, J. Org. Chem., 2005, 70, 4751–4761 CrossRef CAS PubMed . In vinylogous aldol reaction:;
(j) S. Danishefsky, D. F. Harvey, G. Qualich and B. J. Uang, J. Org. Chem., 1984, 49, 393–395 CrossRef;
(k) V. Henryon, L. W. Liu, R. Lopez, J. Prunet and J.-P. Ferezou, Synthesis, 2001, 2401–2414 CAS;
(l) G. Bose and P. Langer, Synlett, 2005, 1021–1023 CAS;
(m) S. E. Denmark and J. R. Heemstra Jr, J. Org. Chem., 2007, 72, 5668–5688 CrossRef CAS PubMed . In bromination:;
(n) J. L. Shamshina and T. S. Snowden, Tetrahedron Lett., 2007, 48(22), 3767–3769 CrossRef CAS;
(o) A. Barbero and F. J. Pulido, Synthesis, 2004, 401–404 CAS.
- Instead of using 7, the addition to aldimines by the dianion generated from the 1,3-diketones was once reported see: J. Jiao, Y. Zhang and R. A. Flowers II, Synlett, 2006, 3355–3357 CAS.
- A. Solladi-Cavallo, C. Marsol, M. Yakoub, K. Azyat, A. Klein, M. Roje, C. Suteu, T. B. Freedman, X. Can and L. A. Nafie, J. Org. Chem., 2003, 68, 7308 CrossRef PubMed.
- The absolute stereochemistry of VMR was determined by comparing spectrum and optical rotation data of known compounds in literature. See: M. J. Niphakis, B. J. Turunen and G. I. Georg, J. Org. Chem., 2010, 75, 6793–6805 CrossRef CAS PubMed . See also ref. 18.
- Hydrogenations on 2,6- disubstituted 2,3-dihydropyridone are known to be stereospecific in literature. For some examples:
(a) D. Ma and H. Sun, Org. Lett., 2000, 2, 2503–2505 CrossRef CAS PubMed;
(b) N. Gouault, M. Le Roch, G. de. C. Pinto and M. David, Org. Biomol. Chem., 2012, 10, 5541–5546 RSC.
-
(a) E. S. Tasber and R. M. Garbaccio, Tetrahedron Lett., 2003, 44, 9185–9188 CrossRef CAS;
(b) E. M. Fry, J. Org. Chem., 1963, 1869–1874 CrossRef CAS.
- J. A. Findlay, W. H. J. Tam and J. Krepinsky, Can. J. Chem., 1978, 56, 613 CrossRef CAS.
- M. W. Edwards, J. W. Daly and C. W. Myer, J. Nat. Prod., 1988, 51, 1188 CrossRef CAS.
- N. Gouault, M. L. Roch, G. C. Pinto and M. David, Org. Biomol. Chem., 2012, 10, 5541–5546 CAS and reference therein.
- For other reported asymmetric synthesis on (+)-241D and/or (−)-241D in literature:
(a) V. H. Vu, F. Louafi, N. Girard, R. Marion, T. Roisnel, V. Dorcet and J.-P. Hurvois, J. Org. Chem., 2014, 79, 3358–3373 CrossRef CAS PubMed;
(b) I. Abrunhosa-Thomas, A. Plas, A. Vogrig, N. Kandepedu, P. Chalard and Y. Troin, J. Org. Chem., 2013, 78, 2511–2526 CrossRef CAS PubMed;
(c) N. Saha and S. K. Chattopadhyay, J. Org. Chem., 2012, 77, 11056–11063 CrossRef CAS PubMed;
(d) N. Gouault, M. Le Roch, G. de. C. Pinto and M. David, Org. Biomol. Chem., 2012, 10, 5541–5546 RSC;
(e) N. Gouault, M. Le Roch, G. de. C. Pinto and M. David, Org. Biomol. Chem., 2012, 10, 5541–5546 RSC;
(f) R. V. N. S. Murali and S. Chandrasekhar, Tetrahedron Lett., 2012, 53, 3467–3470 CrossRef CAS;
(g) K. Damodar and B. Das, Synthesis, 2012, 44, 83–86 CrossRef CAS;
(h) C. K. R. Sateesh, R. G. Venkateswar, G. Shankaraiah, K. Suresh Babu and J. Madhusudana Rao, Tetrahedron Lett., 2010, 51, 1114–1116 CrossRef;
(i) C. Gnamm, C. M. Krauter, K. Broedner and G. Helmchen, Chem.–Eur. J., 2009, 15, 2050–2054 CrossRef CAS PubMed;
(j) J. Monfray, Y. Gelas-Mialhe, J.-C. Gramain and R. Remuson, Tetrahedron: Asymmetry, 2005, 16, 1025–1034 CrossRef CAS;
(k) F. A. Davis, B. Chao and A. Rao, Org. Lett., 2000, 2, 3169–3171 CrossRef;
(l) D. Ma and H. Sun, Org. Lett., 2000, 2, 2503–2505 CrossRef CAS PubMed.
- For some bioactivity studies on isosolenpsin A see:
(a) S. Leclercq, D. Daloze and J. C. Braekman, Org. Prep. Proced. Int., 1996, 28, 499 CrossRef;
(b) G. Howell, J. Butler, R. D. de Shazo, J. M. Farley, H.-L. Liu, N. P. D. Nanayakkara, A. Yates, G. B. Yi and R. W. Rockhold, Ann. Allergy, Asthma, Immunol., 2005, 94, 380–386 CrossRef CAS;
(c) G. B. Yi, D. McClendon, D. Desaiah, J. Goddard, A. Lister, J. Moffitt, R. K. Vander Meer, R. Deshazo, K. S. Lee and R. W. Rockhold, Int. J. Toxicol., 2003, 22, 81–86 CrossRef CAS PubMed.
- For other reported asymmetric synthesis of isosolenpsin A see:
(a) R. S. C. Kumar, E. Sreedhar, G. Venkateswar Reddy, K. Suresh Babu and J. Madhusudana Rao, Tetrahedron: Asymmetry, 2009, 20, 1160–1163 CrossRef CAS;
(b) J. C. Gonzalez-Gomez, F. Foubelo and M. Yus, Synlett, 2008, 2777–2780 CAS;
(c) H. M. T. B. Herath and N. P. D. Nanayakkara, J. Heterocycl. Chem., 2008, 45, 129–136 CrossRef CAS;
(d) N. Girard, J.-P. Hurvois, L. Toupet and C. Moinet, Synth. Commun., 2005, 35, 711–723 CrossRef CAS;
(e) J. Monfray, Y. Gelas-Mialhe, J.-C. Gramain and R. Remuson, Tetrahedron: Asymmetry, 2005, 16, 1025–1034 CrossRef CAS;
(f) S. Ciblat, P. Besse, G. I. Papastergiou, H. Veschambre, J.-L. Canet and Y. Troin, Tetrahedron: Asymmetry, 2000, 11, 2221–2229 CrossRef CAS;
(g) H. Poerwono, K. Higashiyama, T. Yamauchi, H. Kubo, S. Ohmiya and H. Takahashi, Tetrahedron, 1998, 54, 13955–13970 CrossRef CAS;
(h) C. W. Jefford and J. B. Wang, Tetrahedron Lett., 1993, 34, 2911–2914 CrossRef CAS.
-
(a)
A. S. Howard and J. P. Michael, Simple indolizidine and quinolizidine alkaloids, in The Alkaloids, ed. A. Brossi, Academic Press, New York, 1986, pp. 183–308 Search PubMed;
(b)
J. W. Daly, H. M. Garraffo and T. F. Spande, Alkaloids from amphibian skins, in Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, Pergamon Press, Amsterdam, 1999, pp. 1–161 Search PubMed;
(c)
I. Garnett, The quinolizidine alkaloids, in Rodd's Chemistry of Carbon Compounds, ed. M. Sainsbury, Elsevier, Amsterdam, 1998, pp. 181–239 Search PubMed.
-
A. M. Lourenço, P. Máximo, L. M. Ferreira and M. M. A. Pereira, Indolizidine and quinolizidine alkaloids: structure and bioactivity, in Studies in Natural Product Chemistry Bioactive Natural Products Part H, ed. A. Rahman, Elsevier, Amsterdam, 2002, vol. 27, pp. 233–298 Search PubMed.
- As a notable example, quinolizidine alkaloid (−)-cytisine was once identified as a potent partial nicotinic acetylcholine receptor agonist. Inspired by the bioactivity of (−)-cytisine, researchers at Pfizer successfully developed novel smoking cessation drug varenicline approved by FDA in 2006. See J. W. Coe, P. R. Brooks, M. G. Vetelino, M. C. Wirtz, E. P. Arnold, J. Huang, S. B. Sands, T. I. Davis, L. A. Lebel, C. B. Fox, A. Shrikhande, J. H. Heym, E. Schaeffer, H. Rollema, Y. Lu, R. S. Mansbach, L. K. Chambers, C. C. Rovetti, D. W. Schulz, F. D. Tingley and B. T. O'Neill, J. Med. Chem., 2005, 48, 3474 CrossRef CAS PubMed.
-
(a) M. Wink and T. Hartmann, Plant Physiol., 1982, 70, 74–77 CrossRef CAS PubMed;
(b) For a review on quinolizidine alkaloid biosynthesis: S. Bunsupa, M. Yamazaki and K. Saito, Front. Plant Sci., 2012, 3, 239 Search PubMed.
- The stereochemistry results of the reduction on cyclic enaminone were established by literature precedents see:
(a) W. Wysocka and A. Przybyl, Monatsh. Chem., 2001, 132, 973–984 CrossRef CAS;
(b) D. L. Comins, X. Zheng and R. R. Goehring, Org. Lett., 2002, 4, 1611–1613 CrossRef CAS PubMed;
(c) R. T. Yu and T. Rovis, J. Am. Chem. Soc., 2006, 128, 12370–12371 CrossRef CAS PubMed;
(d) T. Nagasaka, H. Yamamoto, H. Hayashi, M. Watanabe and F. Hamaguchi, Heterocycles, 1989, 29, 155–164 CrossRef CAS.
- Similar conditions have also been applied in the region-selective reduction of 2,3-dihydropyridones, M. Dax, R. Frohlich, D. Schepmann and B. Wunsch, Eur. J. Org. Chem., 2008, 6015–6028 Search PubMed.
- For the recent asymmetric synthesis of (−)-epimyrtine see:
(a) T. T. H. Trinh, K. H. Nguyen, P. de A. Amaral and N. Gouault, Beilstein J. Org. Chem., 2013, 9, 2042–2047 CrossRef PubMed;
(b) Y. Ying, H. Kim and J. Hong, Org. Lett., 2011, 13, 796–799 CrossRef CAS PubMed;
(c) S. M. Amorde, A. S. Judd and S. F. Martin, Org. Lett., 2005, 7, 2031–2033 CrossRef CAS PubMed;
(d) F. A. Davis, Y. Zhang and G. Anilkumar, J. Org. Chem., 2003, 68, 8061–8064 CrossRef CAS PubMed;
(e) D. Gardette, Y. Gelas-Mialhe, J.-C. Gramain, B. Perrin and R. Remuson, Tetrahedron: Asymmetry, 1998, 9, 1823–1828 CrossRef CAS.
- D. L. Comins and D. H. LaMunyon, J. Org. Chem., 1992, 57, 5807–5809 CrossRef CAS.
- For other examples on promoting conjugate addition by TMS-Cl:
(a) S. Hanessian and K. Sumi, Synthesis, 1991, 1083–1089 CrossRef CAS;
(b) M. P. Jennings and K. B. Sawant, Eur. J. Org. Chem., 2004, 3201–3204 CrossRef CAS.
- The stereochemistry was established by literature precedents as in ref. 28. See also: J. Thiel, W. Wysocka and W. Boczon, Monatsh. Chem., 1995, 126, 233–239 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra14418j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a