
Metal-free ring expansion of indoles with nitroalkenes: a simple, modular approach to 3-substituted 2-quinolones†
Abstract
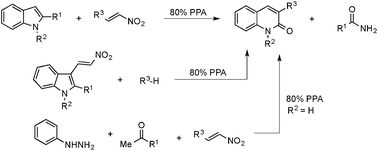
3-Substituted 2-quinolones are obtained via a novel metal-free transannulation reaction of 2-nitroolefins with 2-substituted indoles in polyphosphoric acid. This acid-mediated cascade transformation operates via the ANRORC (Addition of Nucleophile, Ring Opening, and Ring Closure) mechanism and can be used in combination with the Fisher indole synthesis to offer a practical three-component hetero-annulation approach to 2-quinolones from arylhydrazines, 2-nitroalkenes, and acetophenone. An alternative entry to this chemistry employing the alkylation of electron-rich arenes and hetarenes with 1-(2-indolyl)-2-nitroalkene has also been demonstrated.
 Please wait while we load your content...
Please wait while we load your content...

