
Synergetic effect of sodium polystyrene sulfonate and guanidine hydrochloride on the surface properties of lysozyme solutions†
Abstract
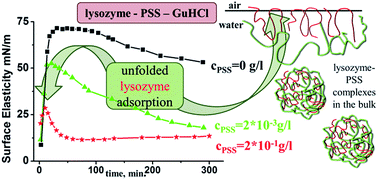
A study of the dilational surface viscoelastic properties of mixed solutions of lysozyme and denaturing agents with different chemical natures allows us to characterize the changes of protein tertiary structure in the surface layer upon adsorption at the liquid–gas interface. We show that guanidine hydrochloride (GuHCl) and urea influence the dynamic surface properties of lysozyme solutions less than the properties of previously studied solutions of bovine serum albumin and β-lactoglobulin. Although the addition of sodium polystyrene sulfonate (PSS) changes the kinetic dependencies of the surface properties and leads to the formation of large aggregates in the bulk phase, the dependencies of the dynamic surface elasticity on the surface pressure almost coincide with the results for pure lysozyme solutions thereby indicating the preservation of the adsorption layer structure. At the same time, the simultaneous addition of PSS and GuHCl to lysozyme solutions results in a strong synergistic effect: the kinetic dependency of the dynamic surface elasticity becomes non-monotonical and similar to that for solutions of amphiphilic polymers and non-globular proteins. The local maximum of this dependency indicates the destruction of the protein globular structure and formation of the distal region of the surface layer. The simultaneous addition of PSS and urea to lysozyme solutions does not lead to a similar effect. These results confirm different mechanisms of protein denaturation under the influence of urea and GuHCl, and the important role of electrostatic interactions in the latter case.
 Please wait while we load your content...
Please wait while we load your content...

