
A ligand-free strategy for the copper-catalysed direct alkynylation of trifluoromethyl ketones†
Lei Wang,
Ning Liu*,
Bin Dai*,
Xiaowei Ma and
Lei Shi
School of Chemistry and Chemical Engineering, Key Laboratory for Green Processing of Chemical Engineering of Xinjiang Bingtuan, Shihezi University, North 4th Road, Shihezi, Xinjiang 832003, China. E-mail: ninglau@163.com; dbinly@126.com; Fax: +86-0993-205-7270; Tel: +86-0993-205-7277
First published on 6th January 2015
Abstract
Most direct alkynylation methods of trifluoromethyl ketones require an additional ligand. In this study, a simple, practical and efficient superbase system for the direct alkynylation of trifluoromethyl ketones was developed without any additional ligand. The catalytic system can tolerate a wide range of functional groups on ketones and alkynes.
The addition of terminal alkynes to trifluoromethyl ketones has attracted considerable interest in the field of organic chemistry, because the method allows the formation of a C–C bond while simultaneously introducing a trifluoromethyl (CF3) group.1 The generated functionalized alkyne products are key building blocks for organic synthesis, especially in the determination of the important uses of efavirenz as a critical anti-HIV drug.2 Organic molecules bearing CF3 groups have received widespread attention in pharmaceutical, agrochemical and organocatalyst fields because of their unique physical and chemical properties.3 Therefore, it is not surprising that different approaches for the construction of such structural unit have been developed (Scheme 1).
 | ||
| Scheme 1 Different methods towards title compounds. | ||
The alkynylation of 2,2,2-trifluoroacetophenone was firstly described using metal acetylides from terminal alkynes and strongly basic organometallic reagents such as Grignard and organolithium reagents (Route A, Scheme 1).4 The addition of trialkylsilylalkynes to trifluoroacetophenone has been also reported (Route B, Scheme 1).5 To access this important class of title compounds, the trifluoromethylation of alkynyl ketones using trifluoromethylation reagents, such as Me3SiCF3 and HCF3, is an alternative strategy (Route C, Scheme 1).6 Recently, chemists' research focus has shifted to the direct alkynylation of trifluoromethyl ketones via transition-metal-catalysed nucleophilic substitution as an economical and ecologically benign alternative (Routes D and E, Scheme 1). Shibasaki and co-workers reported the first example of direct alkynylation of trifluoromethyl ketones through copper/diphosphine or diamine complexes.7 Li et al. reported that simple alkylphosphines, such as tricyclohexylphosphine (PCy3), used in combination with AgF can yield highly active catalysts for the direct alkynylation of trifluoromethyl ketones in water.8 A series of efficient catalysts, such as titanium/chiral cinchona alkaloids,9 copper/N-heterocyclic carbene complexes10 and silver–titania nanocomposites,11 have been successfully applied in alkynylation of trifluoromethyl ketones. However, most methods for the direct alkynylation of trifluoromethyl ketones require an additional ligand.
Although different valuable approaches to the direct alkynylation of trifluoromethyl ketones have been developed, there is an urgent need to search for a simple, general and efficient synthetic protocol for their preparation because of the importance of this class of compounds. Herein, we report a simple and efficient method that requires no additional ligand for the direct alkynylation of trifluoromethyl ketones using commercially available copper salts.
The direct alkynylation of 2,2,2-trifluoroacetophenone with phenylacetylene was selected as a model reaction to optimise the reaction conditions, in which a range of solvent, temperature, base, copper source and atmosphere were investigated.
Firstly, the effect of different solvents on the alkynylation of 2,2,2-trifluoroacetophenone with phenylacetylene was evaluated, and the results showed that solvent served a crucial function in the alkynylation. Dimethyl sulfoxide (DMSO) was most efficient for trifluoroacetophenone conversion and provided a quantitative product yield (ESI, Table S1,† entry 10). The reaction performed in N,N-dimethylformamide (DMF), N,N-dimethylacetamide (DMA), and N-methyl pyrrolidone (NMP) yielded considerably slower conversion (ESI, Table S1,† entries 1–3). We supposed that the strong base of KOtBu combined with high temperature degraded the DMA, DMF, and NMP, and this effect was responsible for the poor results. Therefore, instead of KOtBu, a weak base of K2CO3 was used to optimise the effect of solvents. As expected, DMF, DMA, and NMP provided good results with K2CO3 as base (ESI, Table S1,† entries 11–13). Tetrahydrofuran (THF), toluene, alcohols, and water were also examined as solvents, but only isopropanol was relatively effective for the conversion of trifluoroacetophenone and provided lower product yield (ESI, Table S1,† entries 4–9 and 14–19).
To simplify the operating procedures, control experiments were designed by performing the reaction in air and in nitrogen atmosphere, respectively. The results indicated that the reaction is insensitive to the atmosphere of the reaction condition, and reactions carried out in nitrogen atmosphere exhibited slightly higher catalytic activity than the reactions performed in air (ESI, Table S1,† entry 27). GC-MS analysis showed that oxygen promoted the homo-coupling of phenylacetylene, and an amount of phenylacetylene consumption was responsible for the decrease of efficiency.
In view of the cost, the amount of phenylacetylene was optimised. The reaction rate was not affected when the amount of phenylacetylene was reduced to 1.5 mol equiv., but the rate slowed down in 1.2 mol equiv. of phenylacetylene (ESI, Table S1,† entries 28 and 29). Therefore, using 1.5 mol equiv. of phenylacetylene was the best compromise between cost and product yield (ESI, Table S1,† entry 28).
The effect of reaction temperature on alkynylation was also explored. The reaction smoothly proceeded at 50 °C to 70 °C (Table 1, entries 1 and 2). However, only a trace amount of product was obtained when the temperature was decreased to room temperature (Table 1, entry 3).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | T (°C) | Base | Catalyst | Yieldb (%) |
| a Reaction conditions: 2,2,2-trifluoroacetophenone (0.5 mmol), phenylacetylene (1.0 mmol), Cu source (10 mol%), base (20 mol%) in solvent (1 mL) under N2, 24 h.b Isolated yield.c 1.0 equiv. K2CO3. | |||||
| 1 | DMSO | 70 | KOt-Bu | CuI | 97 |
| 2 | DMSO | 50 | KOt-Bu | CuI | 91 |
| 3 | DMSO | r.t. | KOt-Bu | CuI | Trace |
| 4 | DMSO | 50 | KOH | CuI | 25 |
| 5 | DMSO | 50 | K2CO3 | CuI | 98 |
| 6 | DMSO | 50 | K3PO4 | CuI | 89 |
| 7 | DMSO | 50 | NaOH | CuI | 49 |
| 8 | DMSO | 50 | Na2CO3 | CuI | 60 |
| 9 | DMSO | 50 | NaHCO3 | CuI | 91 |
| 10 | DMSO | 50 | HCOONa | CuI | 90 |
| 11 | DMSO | 50 | NaOt-Bu | CuI | 63 |
| 12 | DMSO | 50 | Cs2CO3 | CuI | 97 |
| 13 | DMSO | 50 | K2CO3 | CuCl | 96 |
| 14 | DMSO | 50 | K2CO3 | Cu2O | 46 |
| 15 | DMSO | 50 | K2CO3 | Cu | 38 |
| 16 | DMSO | 50 | K2CO3 | CuO | 23 |
| 17 | DMSO | 50 | K2CO3 | CuSO4 | 92 |
| 18 | DMSO | 50 | K2CO3 | Cu(NO3)2 | 95 |
| 19 | DMSO | 50 | K2CO3 | Cu(OAc)2 | 99 |
| 20 | DMSO | 50 | None | CuI | Trace |
| 21 | DMSO | 50 | K2CO3 | None | 19 (20c) |
A range of different bases and copper sources was examined for the alkynylation of 2,2,2-trifluoroacetophenone with phenylacetylene using DMSO as a solvent. Among potassium bases, potassium tert-butoxide, carbonate and phosphate were efficient for product formation, with hydroxide providing lower conversion (Table 1, entries 2, 5 and 6 vs. 4). Sodium bicarbonate and sodium formate were superior to other sodium bases (Table 1, entries 9 and 10 vs. 7, 8 and 11). Potassium carbonate and cesium carbonate were the most effective for conversion (Table 1, entry 5 and 12). A series of copper sources [Cu(I), Cu(II) and Cu(0)] was subsequently investigated. Among copper sources in I oxidation states, CuI and CuCl showed considerably higher catalytic activity than Cu2O (Table 1, entries 5 and 13 vs. 14). Copper sources in II oxidation states also exhibited high catalytic activity except for CuO, and Cu(OAc)2 was superior to the other copper sources (Table 1, entries 17–19 vs. 16). Copper powder yielded much slower conversion (Table 1, entry 15). Copper catalysed reactions of alkynes are known to be highly complex and are affected by the oxidation state of the copper source.12 As shown in Table 1, both Cu(I) and Cu(II) works equally well, thereby indicating that copper sources may not act as an alkyne-copper catalyst, but simply as a Lewis acid or enhancer of base solubility. To elucidate the role of copper, a series of Lewis acids instead of copper sources were examined under the same reaction conditions. All tested Lewis acids failed to trigger the reaction (ESI, Table S1,† entries 20–26).
To further explore the role of copper sources, control experiments were designed in the absence of either copper sources or bases. The control experiment revealed that the reaction was difficult to proceed in the presence of CuI but in the absence of bases (Table 1, entry 20). However, 19% product yield was obtained with a base in the absence of copper sources (Table 1, entry 21). A 1.0 equivalent of base in the absence of copper sources was further examined, but 20% product yield was obtained (Table 1, entry 21). These results showed that copper sources promoted reaction rates and served a catalytic function. Early investigation has demonstrated that Cu(II) served as oxidizing agent and could be reduced to Cu(I) by terminal alkynes in the reaction system (Scheme 2).7,13 Observation of the homo-coupling of phenylacetylene is the indirect evidence of Cu(II) reduction. The results showed that Cu(I) is the real oxidation state of copper sources in this catalytic system.
 | ||
| Scheme 2 Process of reducing copper from Cu(II) to Cu(I). | ||
Despite the widespread development of copper-catalysed alkynylation of trifluoromethyl ketones, the mechanism of these processes has remained unclear. Firstly, copper can chelate with carbonyl group of trifluoromethyl ketones to increase the electrophilicity of the carbon atom, while possibly coordinating with alkyne to lower the pKa of terminal hydrogen. To prove which substrate preferentially binds to copper, control experiments have been designed by conducting the reaction in the absence of trifluoroacetophenone or in the absence of phenylacetylene, respectively. Surprisingly, trifluoroacetophenone was full converted to 2,2,2-trifluoro-1-phenylethane-1,1-diol in the presence of Cu(OAc)2 and K2CO3, but in the absence of phenylacetylene (ESI, Fig. S4†).14 2,2,2-Trifluoro-1-phenylethane-1,1-diol is probably an important active intermediate in the catalytic cycle, as reported by Xu.11 However, the terminal hydrogen of phenylacetylene disappeared in presence of Cu(OAc)2 and K2CO3, but in the absence of trifluoroacetophenone (ESI, Fig. S3†). This result indicated that copper may preferentially coordinate with alkyne to form a copper-alkyne complex, which is consistent with previous reports.1c,7
The combination of a strong base and dipolar aprotic solvents such as DMSO, DMF, DMA and NMP, is known to form a superbase media.15 Thus, we hypothesised that the superior performance of DMSO, DMF, DMA and NMP versus other solvents is due to the synergistic effects of the copper catalyst and the superbase media.
To obtain further evidence of the superbase function, the experiment on phenylacetylene H–D exchange was performed. As shown in Fig. 1, the kinetic isotope effect is evident in this catalytic system, thereby indicating that the oxidative addition of copper to alkyne is the rate-determining step. We hypothesised that the superbase promoted the step of oxidative addition of copper to alkyne to generate an active copper-alkyne complex.
 | ||
| Fig. 1 Time course of alkynylation reaction of 2,2,2-trifluoroacetophenone with (a) phenylacetylene and (b) deuterium-modified phenylacetylene. | ||
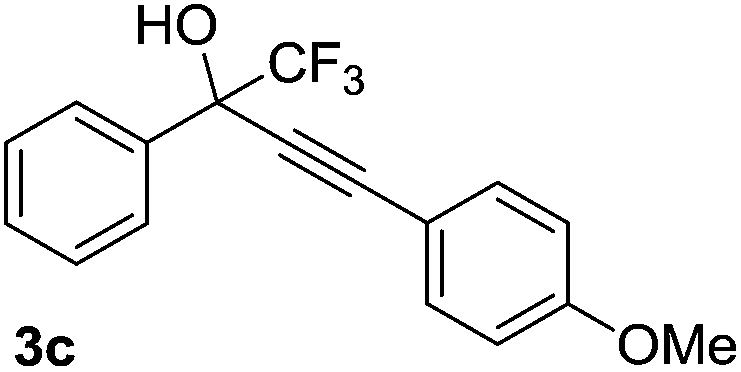
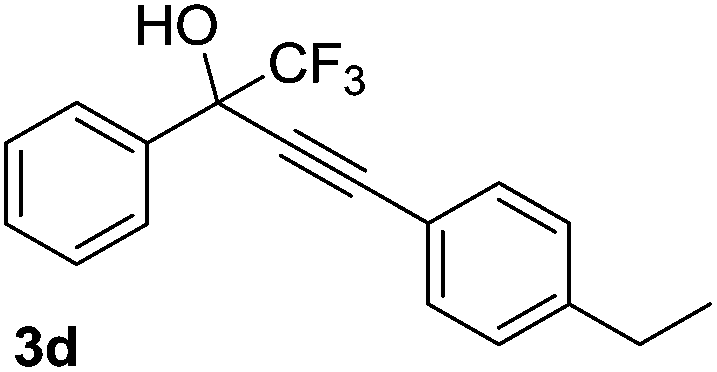
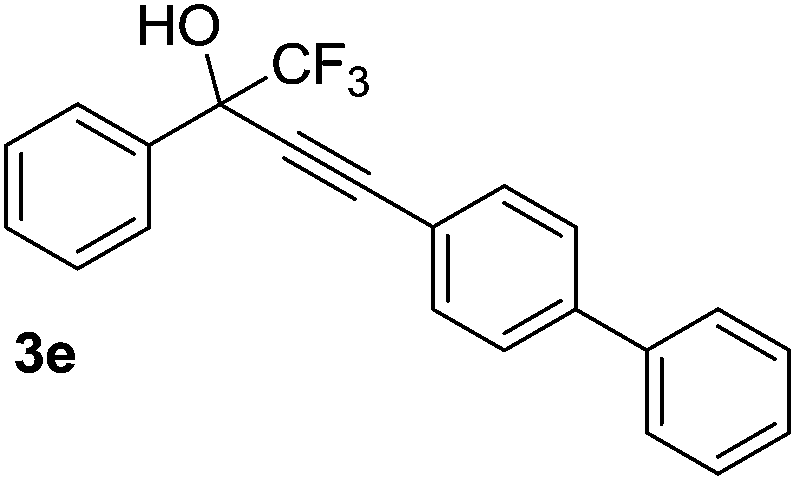
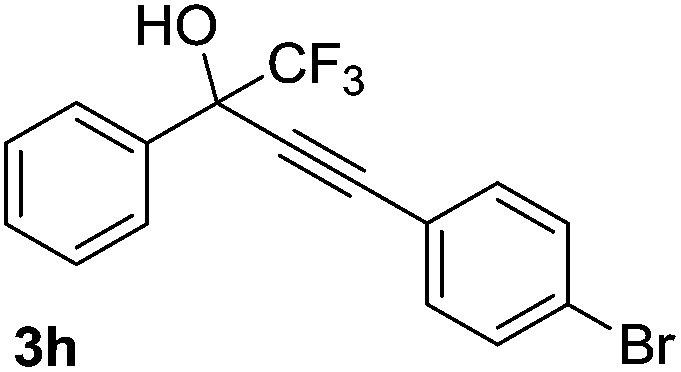
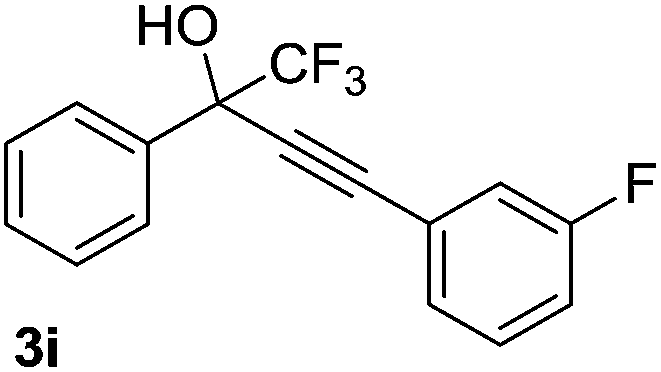
The scope and limitations of addition of the terminal alkynes to trifluoromethyl ketones was examined using Cu(OAc)2 as catalyst, K2CO3 as base and DMSO as solvent under mild reaction conditions. As shown in Table 2, the reactivity of terminal alkynes was tested under optimised reaction conditions, and the desired products were obtained in good-to-excellent yields. The electronic effect of para-substituents bearing the aromatic ring of alkynes was observed. The alkynes containing electron-donating and electron-neutral groups such as 4-Me, 4-OMe, 4-Et, and 4-Ph, reacted smoothly with trifluoroacetophenone to afford the corresponding products in high yields (Table 2, entries 1–5). However, the alkynes containing electron-withdrawing group were less reactive in this system and resulted in moderate product yields (Table 2, entries 6–8). The electronic effect of meta-substituted compounds presented a certain influence on the reaction rate. The reaction worked well with meta-fluorophenyl acetylene bearing the electron-withdrawing group to afford the desired product in good yield (Table 2, entry 9). However meta-methylphenyl acetylene bearing the electron-donating group showed considerably slower conversion at 50 °C, and quantitative product yield was also obtained when the temperature increased to 70 °C (Table 2, entry 10).
|
|||
|---|---|---|---|
| Entry | Alkyne | Product | Yielda (%) |
| a Reaction conditions: ketone (0.5 mmol), alkyne (0.75 mmol), Cu(OAc)2 (10 mol%), K2CO3 (20 mol%) in DMSO (1 mL), 50 °C, 24 h, isolated yield.b 70 °C.c Ten times the amount of the reactants of entries 11 were used, and performed in DMSO (10 mL) at 50 °C for 24 h. | |||
| 1 |  |
 |
96 |
| 2 |  |
 |
91 |
| 3 |  |
 |
96 |
| 4 |  |
 |
89 |
| 5 |  |
 |
92 |
| 6 |  |
 |
87 |
| 7 | 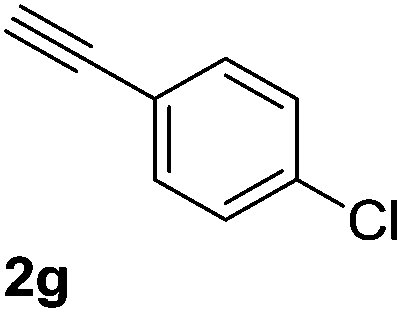 |
 |
65 |
| 8 |  |
 |
77 |
| 9 |  |
 |
63 |
| 10 |  |
 |
5 (97b) |
| 11 |  |
 |
67 |
| 12 |  |
 |
42 (61b) |
| 13 |  |
 |
65 (83b) |
| 14 | 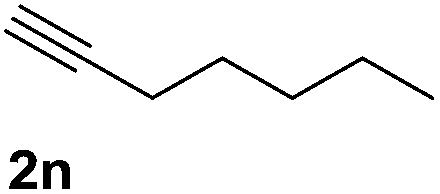 |
 |
28 (71b) |
| 15 |  |
 |
85 |
| 16 | 2k |  |
81 |
| 17 | 2a | 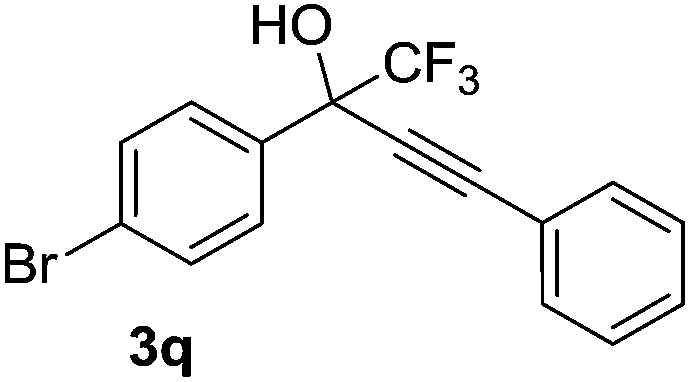 |
93 |
| 18 | 2a |  |
66 |
| 19 | 2a |  |
96 |
| 20c | 2k | 3k | 72 |

To extend the scope of alkynylation, the reaction of alicyclic and aliphatic alkynes was tested. The reactivity of alicyclic and aliphatic alkynes was lower than those of aryl alkynes, and increasing reaction temperature was required to obtain satisfactory results (Table 2, entries 11–14). Moreover, the addition of 3-ethynylthiophene to 2,2,2-trifluoroacetophenone proceeded to 85% product yield in mild reaction conditions (Table 2, entry 15).
The optimised reaction conditions were also applied in the alkynylation of a series of substituted trifluoroacetophenone. The results indicated that alkynylation was evidently affected by the nature of substitution on trifluoroacetophenone. Trifluoroacetophenones bearing electron-withdrawing groups were successfully converted to the desired products in good-to-excellent yields (Table 2, entries 16 and 17). The compounds bearing electron-donating groups resulted in moderate product yield (Table 2, entry 18). Subsequently, with the optimised reaction conditions, the addition of ethyl 3,3,3-trifluoropyruvate with phenylacetylene smoothly proceeded and resulted in quantitative yield (Table 2, entry 19).
The reaction is practical and scalable, and could be scaled up to a gram scale without any problems (Table 2, entry 20).
In conclusion, we successfully developed a general, simple, and efficient method for the direct alkynylation of trifluoromethyl ketones by utilising inexpensive copper salts as pre-catalyst. The major advantages of this process are as follows: no additional ligand is required and the reaction works well in air. A wide range of functional groups on alkynes are compatible with the reaction conditions, including aryl acetylene, alicyclic, and aliphatic alkynes. We hypothesised that the key to the success of the reaction could be the synergistic effects of the copper catalyst and the superbase media. Studies are underway in our laboratory to better elucidate the reaction mechanisms of these processes.
Acknowledgements
The authors thank the financial support from the National Natural Science Foundation of China (21466033), the Start-Up Foundation for Young Scientists of Shihezi University (RCZX201207), the Program for Excellent Talents of Shihezi University (2012ZRKXYQ03), and the National High Technology Research and Development Program of China (2012AA03A611).Notes and references
- (a) C. M. Wei and C.-J. Li, Green Chem., 2002, 4, 39–41 RSC; (b) X. Yao and C.-J. Li, Org. Lett., 2005, 7, 4395–4398 CrossRef CAS PubMed; (c) C.-J. Li, Acc. Chem. Res., 2010, 43, 581–590 CrossRef CAS PubMed; (d) W.-J. Yoo, L. Zhao and C.-J. Li, Aldrichimica Acta, 2011, 44, 43–50 CAS; (e) C. B. Kelly, M. A. Mercadante and N. E. Leadbeater, Chem. Commun., 2013, 49, 11133–11148 RSC.
- (a) R. C. Rizzo, M. Udier-Blagović, D.-P. Wang, E. K. Watkins, M. B. K. Smith, R. H. Smith Jr, J. Tirado-Rives and W. L. Jorgensen, J. Med. Chem., 2002, 45, 2970–2987 CrossRef CAS PubMed; (b) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (d) P. Boonsri, M. Kuno and S. Hannongbua, Med. Chem. Commun., 2011, 2, 1181–1187 RSC; (e) R. Wanke, D. A. Novais, S. G. Harjivan, M. M. Marques and A. M. M. Antunes, Org. Biomol. Chem., 2012, 10, 4554–4561 RSC.
- (a) J. P. Bégué, D. Bonnet-Delpon, B. Crousse and J. Legros, Chem. Soc. Rev., 2005, 34, 562–572 RSC; (b) R. Smits, C. D. Cadicamo, K. Burger and B. Koksch, Chem. Soc. Rev., 2008, 37, 1727–1739 RSC; (c) V. M. Muzalevskiy, A. V. Shastin, E. S. Balenkova, G. Haufe and V. G. Nenajdenko, Synthesis, 2009, 3905–3929 CAS; (d) X. Wang, L. Truesdale and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 3648–3649 CrossRef CAS PubMed; (e) M. A. García-Monforte, S. Martínez-Salvador and B. Menjón, Eur. J. Inorg. Chem., 2012, 4945–4966 CrossRef; (f) S. Dasgupta, K.-W. Huang and J. Wu, Chem. Commun., 2012, 48, 4821–4823 RSC; (g) Z. Weng, W. He, C. Chen, R. Lee, D. Tan, Z. Lai, D. Kong, Y. Yuan and K.-W. Huang, Angew. Chem., Int. Ed., 2013, 52, 1548–1552 CrossRef CAS PubMed; (h) Z. Zhang, Z. Bao and H. Xing, Org. Biomol. Chem., 2014, 12, 3151–3162 RSC; (i) M. Shang, S.-Z. Sun, H.-L. Wang, B. N. Laforteza, H.-X. Dai and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 10439–10442 CrossRef CAS PubMed.
- (a) R. J. Linderman and M. S. Lonikar, J. Org. Chem., 1988, 53, 6013–6022 CrossRef CAS; (b) C. Bittner, A. S. Busemann, U. Griesbach, F. Haunert, W.-R. Krahnert, A. Modi, J. Olschimke and P. L. Steck, Organic Synthesis Workbook II, Wiley-VCH, 2001, pp. 71–84 CrossRef; (c) A. Choudhury, J. R. Moore, M. E. Pierce, J. M. Fortunak, I. Valvis and P. N. Confalone, Org. Process Res. Dev., 2003, 7, 324–328 CrossRef CAS; (d) N. Chinkov, A. Warm and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 2957–2961 CrossRef CAS PubMed.
- (a) R. Motoki, D. Tomita, M. Kanai and M. Shibasaki, Tetrahedron Lett., 2006, 47, 8083–8086 CrossRef CAS PubMed; (b) V. R. Chintareddy, K. Wadhwa and J. G. Verkade, J. Org. Chem., 2011, 76, 4482–4488 CrossRef CAS PubMed.
- (a) H. Kawai, K. Tachi, E. Tokunaga, M. Shiro and N. Shibata, Org. Lett., 2010, 12, 5104–5107 CrossRef CAS PubMed; (b) H. Kawai, T. Kitayama, E. Tokunaga and N. Shibata, Eur. J. Org. Chem., 2011, 5959–5961 CrossRef CAS; (c) H. Kawai, Z. Yuan, E. Tokunaga and N. Shibata, Org. Biomol. Chem., 2013, 11, 1446–1450 RSC.
- R. Motoki, M. Kanai and M. Shibasaki, Org. Lett., 2007, 9, 2997–3000 CrossRef CAS PubMed.
- G.-J. Deng and C.-J. Li, Synlett, 2008, 1571–1573 CAS.
- G.-W. Zhang, W. Meng, H. Ma, J. Nie, W.-Q. Zhang and J.-A. Ma, Angew. Chem., Int. Ed., 2011, 50, 3538–3542 CrossRef CAS PubMed.
- C. A. Correia, D. T. McQuade and P. H. Seeberger, Adv. Synth. Catal., 2013, 355, 3517–3521 CrossRef CAS.
- H. Wang, K.-F. Yang, L. Li, Y. Bai, Z.-J. Zheng, W.-Q. Zhang, Z.-W. Gao and L.-W. Xu, ChemCatChem, 2014, 6, 580–591 CrossRef CAS.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- (a) P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632–2657 CrossRef CAS; (b) Y. S. Zalkind and Fr. B. Fundyler, Ber. Dtsch. Chem. Ges. B, 1936, 69, 128–130 CrossRef; (c) Y. S. Zalkind and B. M. Fundyler, Russ. J. Gen. Chem., 1939, 9, 1725–1728 CAS; (d) A. A. Clifford and W. A. Waters, J. Chem. Soc., 1963, 3056–3062 RSC.
- (a) X. D. Jiang, S. Matsukawa, K. I. Kakuda, Y. Fukuzaki, W. L. Zhao, L. S. Li, H. B. Shen, S. Kojima and Y. Yamamoto, Dalton Trans., 2010, 39, 9823–9829 RSC; (b) N. Duangdee, W. Harnying, G. Rulli, J. M. Neudörfl, H. Gröger and A. Berkessel, J. Am. Chem. Soc., 2012, 134, 11196–11205 CrossRef CAS PubMed; (c) M. von Arx, T. Mallat and A. Baiker, Angew. Chem., Int. Ed., 2001, 40, 2302–2305 CrossRef CAS; (d) C. D. Ritchie, J. Am. Chem. Soc., 1984, 106, 7187–7194 CrossRef CAS.
- (a) Y. Yuan, I. Thomé, S. H. Kim, D. Chen, A. Beyer, J. Bonnamour, E. Zuidema, S. Chang and C. Bolm, Adv. Synth. Catal., 2010, 352, 2892–2898 CrossRef CAS; (b) P. Caubère, Chem. Rev., 1993, 93, 2317–2334 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra14322a |
| This journal is © The Royal Society of Chemistry 2015 |