
Direct extraction of carbonyl from waste polycarbonate with amines under environmentally friendly conditions: scope of waste polycarbonate as a carbonylating agent in organic synthesis†
 b
and
b
and
Abstract
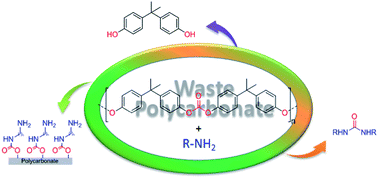
An efficient green method for converting waste polycarbonate into urea derivatives by reacting with primary amines has been developed. Simple treatment of polycarbonate plastic with primary amines in a closed vial at 80 °C without using any catalyst and toxic solvents made this process environmentally friendly. Digestion of the waste polycarbonate obtained from CDs and DVDs with amines affords functionalized urea and 4,4′-(propane-2,2-diyl)diphenol (BPA, bisphenol-A). The procedure is optimized to get maximum conversion of polymer to urea and its derivatives as a major product. The purification procedure to isolate the urea derivatives in the presence of bisphenol-A has been tuned to avoid chromatographic procedures. This environmentally friendly method provides (i) an alternative for recycling BPA from polycarbonate, (ii) a method of obtaining useful product like urea derivatives, (iii) scope for new carbonylating agents in organic synthesis, (iv) an amine functionalized polycarbonate surface.
 Please wait while we load your content...
Please wait while we load your content...

