
Gas-phase dehydration of glycerol over thermally-stable SAPO-40 catalyst†
J. P. Lourenço*a,
A. Fernandesb,
R. A. Bértolob and
M. F. Ribeirob
aCIQA – Centro de Investigação em Química do Algarve, Universidade do Algarve, Campus de Gambelas, 8005-139 Faro, Portugal. E-mail: jlouren@ualg.pt; Fax: +351 289 800 066
bIBB – Instituto de Biotecnologia e Bioengenharia, Instituto Superior Técnico Av. Rovisco Pais, 1049-001 Lisboa, Portugal
First published on 7th January 2015
Abstract
SAPO-40 was used as a catalyst for the gas-phase dehydration of glycerol towards acrolein. At 350 °C the catalyst attained full conversion of glycerol with a negligible deactivation in the first 48 h, a glycerol conversion above 50% after 120 h on stream and a nearly constant selectivity to acrolein above 70%. This catalyst proved to be highly resistant under the experimental conditions used and can be regenerated without loss of activity or significant structural damage. The comparison of SAPO-40 with SAPO-34 and SAPO-11 illustrates the importance of the porous structure and emphasizes the good catalytic performance of this material.
1. Introduction
In the last years there has been an increased demand for fuels and chemicals with less environmental impact than those obtained from fossil resources. Significant efforts have been made to use biomass to achieve this goal, with special focus on biodiesel production. This biofuel is usually produced by transesterification of triglycerides from vegetable oils such as soybean, rapeseed, sunflower and jatropha oils. This process gives rise to a significant amount of glycerol as a by-product. Although glycerol is used in several industries (cosmetics and pharmaceuticals, food additives, coatings), biodiesel production growth will lead to an accumulation of this compound in the near future. The transformation of glycerol into more valuable and versatile feedstocks is therefore of paramount importance to improve the efficiency and economics of biodiesel manufacture.1–3An attractive route for the conversion of glycerol is the acid-catalyzed dehydration to acrolein, which is an important intermediate in the production of acrylic acid, polyesters, superabsorber polymers and detergents.1–5 Currently acrolein is produced by the oxidation of propylene over bismuth molybdate-based catalysts;4 however its production from glycerol may become economically appealing in the near future.
The selective dehydration of glycerol to acrolein has widely been investigated both in gas and liquid-phase,2 with a great variety of catalysts. Nevertheless, conducting the reaction in the liquid-phase, frequently with acids, gives rise to a corrosive medium that makes difficult commercial implementation due to high equipment investments and maintenance costs, catalyst/reaction mixture separation and waste management.2–7 Different types of catalysts have been tested in the dehydration of glycerol in gas-phase, including zeolites,4–11 heteropolyacids4,12–14 metal oxides4,15–17 and modified mesoporous SBA-15 materials.18 This reaction is particularly dependent on the textural and acidic properties of the catalysts. It is generally accepted that the presence of large pores in the catalyst or catalyst support decreases the constraints to the mass transfer and increase the catalytic performance,4,12,18 although interesting results have also been reported with medium pore zeolites such as ZSM-5.8,9 The main products from the glycerol dehydration over acid catalysts, acrolein and acetol, depend on the nature and strength of the acid sites.9,19 The formation of acrolein is favorable over medium to strong acid sites, however the strongest acid sites also promote the formation of coke more effectively, which may lead to a faster deactivation of the catalyst.19
Raw glycerol usually contains water. Thus, to avoid an expensive drying process, it is advantageous to use an acidic catalyst with a high water tolerance, in order to maintain the structural integrity for a longer time in reaction conditions and eventually during a regeneration procedure.
Silicoaluminophosphate (SAPO) family comprises a great variety of structures, usually with mild acidity and variable thermal and hydrothermal stability. SAPO-34, with a cage-like structure with 8-membered ring openings (3.8 Å) and SAPO-40, with a large pore structure involving interconnected 12-membered (6.7 Å) and 8-membered ring (3.7 Å) channels, proved to be highly resistant to thermal and hydrothermal treatments.20–22 Due to the structure properties and the weak to mild acid strength, SAPO-34 has been intensively studied in the dehydration of methanol and is the catalyst in the UOP/Hydro MTO process.23 SAPO-11, with a 10-membered ring (6.5 × 4.0 Å) channels structure is another material of the SAPO family that has found commercial application as catalyst in the Chevron Isodewaxing process.24 W. Suprun et al.25 tested a sample of SAPO-11 and a sample of SAPO-34 in the dehydration of glycerol and found that SAPO-34 is more prone to deactivate than SAPO-11 due to the different pores structure and acidity. In the conditions used, both catalysts showed a significant deactivation rate and a very high selectivity to acetol with increasing time on stream.
In this work, the catalytic performance of SAPO-40 towards the glycerol dehydration is evaluated and compared with the small pore SAPO-34 and medium pore SAPO-11 catalysts.
2. Experimental
2.1. Synthesis of catalysts
SAPO-40 was synthesized according to N. Dumont et al.26 using pseudobohemite (Pural SB) as aluminium source, fumed silica (Cab-OSil M5, Fluka) as silicon source, ortho-phosphoric acid (85% Merck) as phosphorus source and tetrapropylammonium hydroxide (40% aq. sol., Alfa) as templating agent. The final gel with a molar composition of 0.93Al2O3/0.93P2O5/0.65SiO2/2TPAOH/74H2O was submitted to a hydrothermal treatment at 200 °C during 144 h.SAPO-34 was obtained using the original recipe from Fernandes et al.27 The following reagents were used as received: alumina (Pural SB), ortho-phosphoric acid (Merck, 85 wt% aq. solution), silica (Degussa, Aerosil A380), methylamine (Fluka, 41 wt% aq. solution) and tetraethylammonium hydroxide (Alfa 40 wt% aq. solution). The final gel with a molar composition of Al2O3/P2O5/SiO2/TEAOH/0.6MA/40H2O was submitted to a hydrothermal treatment at 170 °C during 24 h.
SAPO-11 used in this study is from the same batch of that published by Bértolo et al.,28 who used alumina (Pural SB), ortho-phosphoric acid (Merck 85 wt% aq. solution), silica (Ludox AS40), MA (Fluka 41 wt% aq. solution) and dipropylamine (Aldrich, 99 wt% aq. solution) as reagents. The gel with a final molar composition of Al2O3/P2O5/0.4SiO2/1.5DPA/50H2O was then submitted to a hydrothermal treatment at 200 °C for 24 h.
2.2. Characterization
The structure type and crystallinity of all the samples were checked by powder X-ray diffraction on a Panalytical X'Pert Pro diffractometer using Cu Kα radiation filtered by Ni and an X'Celerator detector. 29Si MAS NMR spectra were recorded on a Bruker Avance 400 at 79.5 MHz, using 40° pulses, a spinning rate of 5.5 kHz and a recycle delay of 12 s. SEM micrographs were obtained on a JEOL JSM-7001F equipment. Chemical composition of the various samples was determined by elemental chemical analysis (ICP for Al and P and AAS for Si). Pyridine (Py) adsorption was followed by FTIR spectroscopy using a home-made Quartz cell allowing sample vacuum and temperature pretreatment and subsequent Py adsorption at 150 °C. Quantitative measurements were done as described elsewhere.28 The results are listed in Table 1. Thermogravimetric data (TG) were obtained with a TG92 Setaram apparatus. Nitrogen sorption experiments were performed using an ASAP 2010 series equipment from Micromeritics. Prior to measurements, samples were outgassed first at 90 and then at 350 °C, during 1 and 4 hours, respectively.| Sample | Chemical composition (TO2) | Acid sites (μmol g−1) | Textural properties | ||
|---|---|---|---|---|---|
| Lewis | Brönsted | ABET (m2 g−1) | Vmicrop.b (cm3 g−1) | ||
| a As determined in ref. 28.b Determined by t-plot method, nd – not determined. | |||||
| SAPO-40 | (Si0.12Al0.47P0.41)O2 | 56 | 280 | 670 | 0.27 |
| SAPO-11 | (Si0.06Al0.50P0.44)O2 | 49a | 46a | 210 | 0.07 |
| SAPO-34 | (Si0.17Al0.48P0.35)O2 | nd | nd | 640 | 0.27 |
2.3. Catalytic tests
The dehydration of glycerol was performed at different temperatures in the range 320–380 °C under atmospheric pressure, in a fixed-bed vertical quartz reactor (1.5 cm i.d.) using 300 mg of catalyst. The temperature was controlled by a thermocouple placed near the catalyst bed. Before each test, the catalyst was heated at 500 °C under a flux of dry nitrogen (30 mL min−1) and kept during 2 h. The reaction feed, an aqueous solution containing 10, 30 or 50 wt% of glycerol, was introduced in the reactor by a syringe pump KD Scientific at variable WHSV (0.85–4.69 h−1) and diluted in a flow of dry nitrogen (30 mL min−1). The reaction products were collected in a NaCl/ice trap followed by two additional water traps. Reaction products were analyzed on a Chrompack CP2001 gas chromatograph equipped with a 25 m OPTIMA FFAP Macherey Nagel capillary column and a FID detector. For quantitative measurements 1-propanol (for low boiling point products) and 1,4-butanediol (for glycerol) were used as internal standards.The conversion and selectivity were calculated as follows:

where ng,total is the total number of moles of glycerol injected into the reactor during the time on stream t, ng,t the number of moles of glycerol in the products recovered at time on stream t and ng,i,t the number of moles of glycerol converted to the product i during the time on stream t. In order to obtain significant amounts of products, each analysis corresponds to the products usually recovered for 3 h for low space velocities and for 1 h for high space velocities.
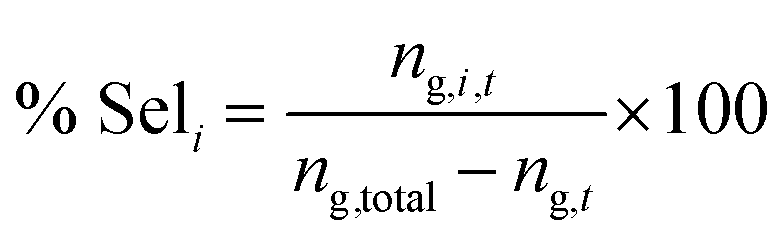
3. Results and discussion
3.1. Synthesis and characterization
Fig. 1 displays the powder XRD patterns of the as-synthesised SAPO-40, SAPO-34 and SAPO-11 materials. The patterns indicate high crystalline solids and only the crystalline phase corresponding to SAPO-40 (AFR), SAPO-34 (CHA) and SAPO-11 (AEL) could be detected in the respective patterns (ICDD PDF2 cards 01-087-1146, 00-055-0829 and 00-041-0023, respectively). SEM micrographs (ESI, Fig. SDF1†) confirm the high purity of the samples. The different structures show the typical crystal shape of these materials and no significant amount of amorphous materials is observed. | ||
| Fig. 1 Powder XRD patterns of the as-synthesized samples. | ||
The acidity of SAPO materials is dependent on both the amount of silicon incorporated and the mechanism of incorporation. Substitution of a P atom in the neutral aluminophosphate framework leads to the formation of Si–O–Al bridges with a negative charge that may be balanced by an acidic proton. This type of incorporation corresponds to mechanism SM2 and gives rise to isolated Si atoms and consequently isolated and weak acid sites. However, depending on the initial amount of silicon in the synthesis gel and other experimental parameters, the incorporation may also occur by mechanism SM3, where two Si atoms substitute a pair of neighbouring P and Al leading to the formation of silicon islands. Due to different silicon environments, the acid sites located at the edges of silicon islands were proved to have higher strength than those resulting from isolated Si atoms.29–32 Therefore, the silicon distribution in the various SAPO-samples is an important feature that plays a key role in the catalytic activity. Fig. 2 displays 29Si NMR spectra of the samples tested. Despite the broad and noisy signals observed (due to the very small amount of Si and the rather low abundance of 29Si nucleus), all the samples show resonance lines in the range −90 to −110 ppm where are usually found the Si(4Al), Si(3Al), Si(2Al), Si(1Al) and Si(4Si) environments.30 Nevertheless, the relative intensities of these resonances are somewhat different for the three samples. SAPO-34 shows a broad and intense peak between −90 and −95 ppm (respectively Si(4Al) and Si(3Al) environments) together with a weaker signal at ca. −110 ppm indicative of a low amount of silica islands. SAPO-40 also shows intense resonance lines between −90 and −95 ppm but with resonance signals centered at ca. −110 ppm with higher intensity than that observed for SAPO-34. This indicates the presence of a significant amount of isolated Si species together with silica domains. SAPO-11 shows a similar profile when compared with SAPO-40 sample, though with a more intense silica islands signal. Although the presence of silicon-rich domains may give rise to strong acid sites, if these domains become excessively large, as suggested by 29Si NMR spectrum of SAPO-11, the fraction of silicon atoms contributing to the acidity would be clearly reduced. This may explain the reduced amount of Brönsted acid sites measured by pyridine adsorption on SAPO-11 when compared with SAPO-40 (Table 1), where the difference of silicon incorporated only partially accounts for the difference in the number of Brönsted acid sites.
 | ||
| Fig. 2 29Si MAS NMR spectra of the different SAPO samples. | ||
3.2. Catalytic activity of SAPO-40
 | ||
| Scheme 1 Pathways for the formation of the main products from dehydration of glycerol. | ||
The influence of the reaction temperature on the catalytic performance of SAPO-40 is illustrated in Fig. 3 and Table 2. At the lowest temperature tested (320 °C) a significant deactivation occurs in the first hours on stream, followed by a gradual stabilization of the catalytic activity. This behavior of the catalytic activity is compatible with a partial blocking of the porous system in early stages of the reaction that may arise from the constraints to the diffusion of glycerol and reaction products imposed by the dimension of the pores and the rather low temperature.
 | ||
| Fig. 3 Conversion of glycerol at 320 °C (▲), 350 °C (■) and 380 (●) (experimental conditions: WHSV of 0.85 h−1, 10 wt% glycerol solution). | ||
In these operating conditions, the conversion at 2 h on stream reaches 90%. This indicates that probably part of the acid sites are not active at that temperature due to a weak acid strength and, therefore, one may anticipate that an increase of the temperature will be advantageous. In fact, increasing the temperature to 350 °C favors the catalytic activity reflected in the increase of the initial conversion and also reduces the diffusional constraints, since no deactivation is observed in the first hours on stream. Nevertheless, a further increase of the temperature to 380 °C causes a higher deactivation of the catalyst. Additionally to the effects on the diffusion of reaction products, the temperature also influences the side reactions that take place and, consequently, the type of coke that is formed. This is well documented for the conversion of hydrocarbons over zeolites33 and usually the coke that is formed at high temperature has a lower H/C ratio (hard-coke) and frequently a higher molecular weight than that formed at low temperature (light-coke). This hard-coke decomposes at higher temperature under oxygen. In the present case, a small increase of the amount of coke is observed with the increase of the temperature (Table 2) but no significant changes can be observed in the TG profile between 150 and 700 °C of the spent catalyst (ESI, Fig. SDF2†), however, the heat profile (ESI, Fig. SDF2†) shows two degradation steps with maxima at ca. 365 °C and 520–550 °C. It is evident from the heat profile that the degradation process occurring at the lower temperature is more important for the catalyst used at 320 °C. On the other hand, the maximum of the high temperature process increases from 520 °C for the catalyst used at 320 °C to 550 °C for the catalyst used at 380 °C. These data suggest the presence of coke entities with higher molecular weight and/or lower H/C and O/C ratios when the reaction is performed at higher temperature. At lower temperature (320 °C) the presence of glycerol-based small oligomers and other oxygenated compounds is certainly more likely. Due to the rather low temperature these oligomers do not diffuse easily out of the pores of the catalyst but at the same time do not evolve rapidly to large molecular weight coke molecules that permanently deactivate the catalyst active sites. This balance is probably one of the main reasons for the conversion profile observed at 320 °C.
Data in Table 2 indicate that for SAPO-40 material the selectivity to acrolein is nearly constant in the temperatures range under study. The comparison of these values with those obtained for other microporous catalysts such as zeolites is not straightforward due to the different experimental conditions employed. Nevertheless, the results reported here for SAPO-40 are similar to some of the best ones reported for zeolitic catalysts: Y. Gu et al.6 tested a nanosized HZSM-11 and reported a selectivity to acrolein of 74.9% for a conversion of 81.6%, at 320 °C and 8 h on stream; Corma et al. found a selectivity to acrolein of 62% for a glycerol conversion of 100% at 350 °C and using ZSM-5 as catalyst;8 using ferrierite at 340 °C and 91.7 mol% H2O in the feed, Kim et al.9 reported a selectivity of 77.1% with a conversion of 70.9% after 2 h reaction time.
The selectivity to acetol and acetaldehyde is for all the cases relatively low, however the production of acetaldehyde is clearly favored as the temperature rises. It has been suggested that acetaldehyde may result from the decomposition of 3-hydroxypropanal, an intermediate in the pathway leading to acrolein, by a retro-aldol condensation and thus limiting the formation of this main product.12
 | ||
| Fig. 4 Conversion of glycerol over SAPO-40 at different WHSV: 0.86 h−1 (□), 2.68 h−1 (◇) and 4.69 h−1 (○) (experimental conditions: 350 °C, 30 wt% glycerol solution). | ||
For the lowest WHSV tested no significant deactivation is observed for the first hours on stream. On the other hand, increasing the space velocity a faster pore blockage may be expected and a significant deactivation in the first hours on stream is now observed.
Table 3 shows the selectivity for the major reaction products. These data reflect not only the increase of the glycerol space velocity but also the increase of water–catalyst ratio, because using a constant ratio water–glycerol (30 wt% aq. sol.) leads to the increase of both parameters. The amount of water in the feed influences the selectivity as shown by Y. T. Kim et al.,9 using a constant concentration of glycerol. A high amount of water competing with glycerol for adsorption on the active sites may suppress side reactions such as bimolecular condensations that lead to the formation of large molecules and thus increase the selectivity to acrolein. Even if the amount of glycerol in the feed also increases as in the present case, the same effect may probably be expected. Therefore, the data in Table 3 should reflect not only the increase of the glycerol space velocity but also the increase of water–catalyst ratio. Indeed, when comparing the selectivity to acrolein at WHSV of 0.86 and 2.68 h−1 a decrease could be expected9,34 probably due to the higher concentration of glycerol, intermediates and reaction products that favors side reactions such as bimolecular condensations. However this effect is compensated by the increase of the water in the feed (1.9 mL per h per g catalyst and 5.8 mL per h per g catalyst, respectively) and thus an increase of the selectivity to acrolein is observed. Comparing the results of Table 2 (350 °C, WHSV of 0.85 h−1, water flow of 7.5 mL per h per g catalyst) with those of Table 3 (350 °C, WHSV of 0.86 h−1, water flow of 1.9 mL per h per g catalyst), this effect is more pronounced and a substantial increase of the initial selectivity to acrolein is obtained for total glycerol conversion.
| WHSV (h−1) | 0.86 | 2.68 | 4.69 |
|---|---|---|---|
| Conv. (%) | 100 | 100 | 79 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Select. (%) | |||
| Acrolein | 63.7 | 70.0 | 68.1 |
| Acetol | 4.3 | 7.9 | 10.4 |
| Acetaldehyde | 11.7 | 8.7 | 6.2 |
| Time on stream | 1 cycle | 2 cycle | ||
|---|---|---|---|---|
| 2.5 h | 120 h | 2.5 h | 120 h | |
| Conv. (%) | 100 | 54 | 100 | 61 |
|
||||
| Select. (%) | ||||
| Acrolein | 75.7 | 73.0 | 72.8 | 74.2 |
| Acetol | 5.7 | 8.0 | 6.4 | 8.2 |
| Acetaldehyde | 13.7 | 7.8 | 12.2 | 10.2 |
A regeneration of the spent catalyst under a flux of dry air at 530 °C for 12 h leads to a catalyst very similar to the original both in terms of conversion and selectivity, as shown in Fig. 5 and Table 4. The integrity of the structure after two regeneration cycles is also clearly evidenced by the high quality powder XRD pattern obtained after a second regeneration (ESI, Fig. SDF3†).
 | ||
| Fig. 5 Conversion of glycerol over SAPO-40. Fresh catalyst (■) and regenerated catalyst (◆) (experimental conditions: WHSV of 0.85 h−1, 10 wt% glycerol solution). | ||
Fig. 6 displays the catalytic activity of the various SAPOs, tested at 350 °C using 10 wt% glycerol. In these conditions SAPO-34 suffers a strong deactivation in the first 24 h on stream. On the other hand, SAPO-40 retains a very high activity for 48 h, followed by a gradual deactivation. The behavior of SAPO-34 could, in some extent, be anticipated due to the particular pore structure, i.e. large cages connected by small pores with openings of 3.8 Å. It is worth noting that the microporous volume measured for SAPO-34 and SAPO-40 is the same (Table 1), however the cage openings of SAPO-34 are much smaller than the pores of SAPO-40. In this case, the significant deactivation observed is probably due to the easy blocking of the porous system by coke formation (see Table 5). The remaining activity probably results from acid sites located at the external surface of the crystals. The amount of coke after 120 h on stream found for SAPO-34 is significantly lower than that found for SAPO-40 (Table 5), which agrees with a more efficient pore blockage. SAPO-11 shows an intermediate behavior, which is in line with the type of the porous structure with 10-membered ring pore diameter. In this case, the formation of coke does not promote a pore blockage as efficiently as in SAPO-34, since a similar amount of coke formed does not give rise to the same deactivation rate. When compared with SAPO-34, the larger pore diameter would give rise to fewer constraints; however the lower microporous volume (Table 1) and the one-dimensional circulation of reagents and products have the opposite effect and thus the combined result of the two factors is observed. A similar deactivation behavior of SAPO-34 and SAPO-11 was already reported by W. Suprun et al.25 When compared with the other SAPO structures, SAPO-40 shows a remarkable catalytic stability, which certainly results from the more open structure and a more adequate acidity. In fact, the high amount of silicon incorporated and the non-excessively large silica domains existing in SAPO-40 sample, as evidenced by 29Si NMR spectra, provide a large number of acid sites when compared with SAPO-11 (Table 1).
 | ||
| Fig. 6 Conversion of glycerol over SAPO-34 (◆), SAPO-11 (▲) and SAPO-40 (■) (experimental conditions: WHSV of 0.85 h−1, 10 wt% glycerol solution). | ||
| Time on stream | SAPO-34 | SAPO-11 | SAPO-40 | |||
|---|---|---|---|---|---|---|
| 2.5 h | 120 h | 2.5 h | 120 h | 2.5 h | 120 h | |
| a Weight loss by TG in the range 150–700 °C. | ||||||
| Conv. (%) | 90 | 16 | 100 | 26 | 100 | 54 |
|
||||||
| Select. (%) | ||||||
| Acrolein | 76.3 | 72.2 | 73.0 | 74.7 | 75.7 | 73.0 |
| Acetol | 10.4 | 11.5 | 10.5 | 9.9 | 5.7 | 8.0 |
| Acetald. | 8.1 | 10.9 | 9.1 | 8.7 | 13.7 | 7.8 |
| Propion. | 1.5 | 3.0 | 1.7 | 0.4 | 2.7 | 1.3 |
| Cokea (%) | 9.9 | 9.8 | 20.1 | |||
For all the samples under study, the main products obtained are acrolein, acetol, acetaldehyde, and, in lower amount, propionaldehyde (Table 5 and ESI, Fig. SDF4–SDF6†). In spite of the different deactivation rates, all the materials show a high selectivity to acrolein during the catalytic test. These data suggest that the acidity of the different materials, regardless of the variations due to the difference in silicon distribution that may influence the coke formation and consequently the deactivation rate, is in all the cases adequate for the formation of acrolein in the present conditions. The low selectivity for acetol, when compared with the selectivity to acrolein for all the samples, indicates that the majority of the active sites are the Brönsted ones.33 This is particularly evident in the case of SAPO-11 where the number of Lewis acid sites is higher than the number of Brönsted sites (Table 1), but the selectivity obtained clearly suggests the predominance of Brönsted sites. Although the acidity of SAPO-34 cannot be assessed using pyridine,35 from the silicon distribution evidenced by 29Si NMR it is expected that SAPO-34, with a low amount of silica domains, comprises mainly weak Brönsted acid sites where the presence of silica domains in the other materials may give rise to Brönsted acid sites with higher strength.22,29–32 Nevertheless, the similar selectivity obtained for all the catalysts suggests that even the weak Brönsted acid sites of SAPO-34 seem to be active for this reaction in the experimental conditions used in this study. For the SAPOs tested, no particular trend was observed in the selectivity with the number or strength of acid sites.
The selectivity found for SAPO-11 and SAPO-34 do not agree with the findings of Suprun et al.25 who reported a very high selectivity to acetol and a significant decrease of the selectivity to acrolein with time on stream. It should be noted, however, that the results are hardly comparable since the composition and silicon distribution of the catalysts used by these authors was not reported and, additionally, the catalytic tests were performed in different conditions.
4. Conclusions
Silicoaluminophosphate SAPO-40 proved to be a highly active and stable catalyst for the dehydration of glycerol towards acrolein. The highest glycerol conversion and acrolein selectivity combined with the highest stability were obtained for a temperature of 350 °C, which should reflect the balance of the effect of the constraints to the mass transfer imposed by the structure and the strength of the active sites.From the data obtained it was clearly seen that SAPO-40 can be easily regenerated and does not show significant structural damage after two regeneration cycles.
The different silicoaluminophosphate catalysts evaluated in this study show similar catalytic performance for short times on stream, which probably reflects the same nature of the acid sites, whereas for longer times on stream the effect of the pore structure is emphasized. SAPO-34 with a porous structure involving small pore openings shows a significant deactivation in the first hours on stream, which contrasts with SAPO-40 with a more open one. SAPO-11 exhibits an intermediate behavior.
Acknowledgements
The authors thank Prof. João Rocha (CICECO, Aveiro, Portugal) for providing the NMR spectra and Portuguese FCT for financial support (SFRH/BD/73234/2010 and SFRH/BPD/91397/2012).References
- L. Liu, X. P. Ye and J. J. Bozell, ChemSusChem, 2012, 5, 1162 CrossRef CAS PubMed.
- B. Katryniok, S. Paul, M. Capron and F. Dumeignil, ChemSusChem, 2009, 2, 719 CrossRef CAS PubMed.
- N. Rahmat, A. Z. Abdullah and A. R. Mohamed, Renewable Sustainable Energy Rev., 2010, 14, 987 CrossRef CAS PubMed.
- B. Katryniok, S. Paul, V. Belliere-Baca, P. Reye and F. Dumeignil, Green Chem., 2010, 12, 2079 RSC.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. D. Pina, Angew. Chem., Int. Ed., 2007, 46, 4434 CrossRef CAS PubMed.
- Y. Gu, N. Cui, Q. Yu, C. Li and Q. Cui, Appl. Catal., A, 2012, 429–430, 9 CrossRef CAS PubMed.
- Y. T. Kim, K.-D. Jung and E. D. Park, Microporous Mesoporous Mater., 2010, 131, 28 CrossRef CAS PubMed.
- A. Corma, G. W. Huber, L. Sauvanaud and P. O'Connor, J. Catal., 2008, 257, 163 CrossRef CAS PubMed.
- Y. T. Kim, K.-D. Jung and E. D. Park, Appl. Catal., A, 2011, 393, 275 CrossRef CAS PubMed.
- L. G. Possato, R. N. Diniz, T. Garetto, S. H. Pulcinelli, C. V. Santilli and L. Martins, J. Catal., 2013, 300, 102 CrossRef CAS PubMed.
- C. S. Carriço, F. T. Cruz, M. B. Santos, H. O. Pastore, H. M. C. Andrade and A. J. S. Mascarenhas, Microporous Mesoporous Mater., 2013, 181, 74 CrossRef PubMed.
- E. Tsukuda, S. Sato, R. Takahashi and T. Sodesawa, Catal. Commun., 2007, 8, 1349 CrossRef CAS PubMed.
- H. Atia, U. Armbrusterb and A. Martin, J. Catal., 2008, 258, 71 CrossRef CAS PubMed.
- A. Alhanash, E. F. Kozhevnikova and I. V. Kozhevnikov, Appl. Catal., A, 2010, 378, 11 CrossRef CAS PubMed.
- P. Lauriol-Garbay, J. M. M. Millet, S. Loridant, V. Bellière-Baca and P. Rey, J. Catal., 2011, 280, 68 CrossRef CAS PubMed.
- M. Massa, A. Andersson, E. Finocchio, G. Busca, F. Lenrick and L. Reine Wallenberg, J. Catal., 2013, 297, 93 CrossRef CAS PubMed.
- S.-H. Chai, H.-P. Wang, Y. Liang and B.-Q. Xu, J. Catal., 2007, 250, 342 CrossRef CAS PubMed.
- J. P. Lourenço, M. I. Macedo and A. Fernandes, Catal. Commun., 2012, 19, 105 CrossRef PubMed.
- S.-H. Chai, H.-P. Wang, Y. Liang and B.-Q. Xu, Green Chem., 2007, 9, 1130 RSC.
- C. Minchev, Y. Neiska, V. Valtchev, V. Minkov, T. Tsoncheva, V. Panchev, H. Lechert and M. Hess, Catal. Lett., 1993, 18, 125 CrossRef.
- J. P. Lourenço, M. F. Ribeiro, F. R. Ribeiro, J. Rocha, Z. Gabelica and E. G. Derouane, Microporous Mater., 1995, 4, 445 CrossRef.
- J. P. Lourenço, M. F. Ribeiro, F. R. Ribeiro, J. Rocha and Z. Gabelica, Appl. Catal., A, 1996, 148, 167 CrossRef.
- G. A. Funk, D. Myers and B. Vora, Hydrocarbon Engineering, December 2013, p. 25 Search PubMed.
- M. Rigutto, R. van Veen and L. Huve, Stud. Surf. Sci. Catal., 2007, 168, 855 CrossRef.
- W. Suprun, M. Lutecki, T. Haber and H. Papp, J. Mol. Catal. A: Chem., 2009, 309, 71 CrossRef CAS PubMed.
- N. Dumont, Z. Gabelica, E. G. Derouane and F. Di Renzo, Microporous Mater., 1994, 3, 71 CrossRef CAS.
- A. Fernandes, F. Ribeiro, J. P. Lourenço and Z. Gabelica, Stud. Surf. Sci. Catal., 2008, 174, 281 CrossRef.
- R. Bértolo, J. M. Silva, F. Ribeiro, F. J. Maldonar-Hódar, A. Fernandes and A. Martins, Appl. Catal., A, 2014, 485, 230 CrossRef PubMed.
- D. Bartohomeuf, Zeolites, 1994, 14, 394 CrossRef.
- J. A. Martens, P. J. Grobet and P. A. Jacobs, J. Catal., 1990, 126, 299 CrossRef CAS.
- J. P. Lourenço, M. F. Ribeiro, F. R. Ribeiro, J. Rocha, Z. Gabelica, N. Dumont and E. G. Derouane, Stud. Surf. Sci. Catal., 1994, 84, 867 CrossRef.
- G. Sastre, D. W. Lewis and C. R. A. Catlow, J. Phys. Chem., 1997, 101, 5249 CrossRef CAS.
- F. Bauer and H. G. Karge, in Molecular Sieves, ed. H. G. Karge and J. Weitkamp, Springer-Verlag, Berlin, 2007, vol. 5, p. 249 Search PubMed.
- B. O. Dalla Costa, M. A. Peralta and C. A. Querini, Appl. Catal., A, 2014, 472, 53 CrossRef CAS PubMed.
- R. B. Borade and A. Clearfield, J. Mol. Catal., 1994, 88, 249 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra14241a |
| This journal is © The Royal Society of Chemistry 2015 |