
Superior nano-mechanical properties of reduced graphene oxide reinforced polyurethane composites†
Tejendra K. Guptaa,
Bhanu P. Singh*a,
Ravi Kant Tripathic,
Sanjay R. Dhakate*a,
Vidya N. Singhb,
O. S. Panwarc and
Rakesh B. Mathura
aPhysics and Engineering of Carbon, Division of Materials Physics and Engineering, CSIR-National Physical Laboratory, New Delhi-110012, India. E-mail: bps@nplindia.org; dhakate@nplindia.org; Fax: +91-11-45609310; Tel: +91-11-45608460
bElectron and Ion Microscopy Section, CSIR-National Physical Laboratory, New Delhi-110012, India
cPolymorphic Carbon Thin Films Group, Physics of Energy Harvesting, CSIR-National Physical Laboratory, New Delhi-110012, India
First published on 29th January 2015
Abstract
Polyurethane (PU) based composites were prepared by solvent casting techniques using different wt% (0–5 wt%) of reduced graphene oxide (RGO) as reinforcement. A nanoindentation study has been carried out on these composite sheets in order to investigate their nano-mechanical properties. Incorporation of different wt% RGO into the PU matrix led to a significant increase in the hardness and elastic modulus of the composites. The maximum nanoindentation hardness of 140 MPa for 5.0 wt% RGO loading was observed as compared to 58.5 MPa for pure PU (an overall improvement of 139%). The nanoindentation elastic modulus for the 5.0 wt% RGO loaded sample was 881.7 MPa as compared to 385.7 MPa for pure PU (an overall improvement of 129%). The enhancement in the nano-mechanical properties was correlated with spectroscopic and microscopic investigations using Raman spectroscopy, scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Due to their excellent nano-mechanical properties, these composites find usefulness in structural applications such as the automobile and wind mill blade industries. These composites can also be used in hard and scratch-less coatings on automotive vehicles. The experimental results were in good agreement with theoretical predictions.
1. Introduction
The improved mechanical properties of polymer composites might be a key criterion for the durability of these materials in tribological applications. The mechanical properties of polymer composites are important parameters in the tribological configuration process. In the past few decades, the use of inorganic nanomaterials as reinforcements in the preparation of conducting polymer/inorganic nanocomposites has attracted increasing interest due to their unique properties and various potential applications in automotive, aerospace, antistatic coatings, corrosion resistant coating, construction and electronic industries.1–3Nowadays, stretchable (high strain to failure) conducting materials have received increasing attention because of their attractive applications such as; smart clothing, flexible displays, EMI shielding and electronic textiles.4–6 Shape memory polymers are in high demand due to their stretchable and flexible behavior. Shape memory polymer materials that are used as smart materials, the amount of recovery deformation are particularly large in shape memory polymer and its practical application is widely expected.7 Therefore, polyurethane (PU) is one of the most adaptable thermoplastic elastomeric shape memory polymer materials8–10 which consists of linear segmented block copolymers composed of hard and soft segments.11 It has many useful features, including elasticity, transparency, good abrasion resistance, good chemical resistance, wear resistance, good weather resistance and besides it these composites also have good mechanical properties.12
This work reports the preparation, characterization and measurement of the nano-mechanical properties of PU composites with reduced graphene oxide and explains the structure property connections of the resulting composites with a perspective to enhance its nano-mechanical properties.
Carbon nanotubes (CNTs) are well known as an ultimate nanofiller to enhance the mechanical,13,14 electrical15 and thermal properties16 of polymer composites. However, the inherent bundling nature of CNTs, high cost, poor dispersibility and intrinsic impurities have made them less attractive for practical applications.17 Very recently, the role of CNTs in polymer composites is strongly challenged by much cheaper graphene sheets, which have similar properties like CNTs.18 This is due to the fact that synthesis of CNT is costly as compared to graphene and CNTs have no functionality to make strong bonding with polymer matrix. Therefore, to create some functionality in CNTs, functionalization is necessary. Reduced graphene oxide (RGO) synthesized by chemical methods have large number of edge and basal plane functional groups and these functional group can make strong bonding with polymer matrix to enhance the mechanical properties of the polymer composites.
Two commonly known issues need to be considered in the formation of polymer composites with strong mechanical performance. One is homogeneous dispersion of nanofillers in polymeric matrix and other is a strong interface between nanofillers and polymeric matrix. Therefore, RGO is appropriate nanofiller having oxygenated functional groups which can provide not only better dispersion but also active sites to form ideal interface between RGO and polymer matrix via strong chemical bonding. PU is a suitable polymer that can form strong chemical bonding with RGO via reaction between isocyanate groups at the end of PU chains and oxygenated groups on the RGO.
The focus of the present research is to fabricate RGO reinforced PU matrix composites and to study the nano-mechanical properties by varying reinforcement loading. PU is one of today's most versatile industrial polymers which has been extensively used as surface coatings for various substrates and RGO is able to make PU based scratchless surface coatings material much stronger and more protective.
There are several reports on graphene reinforced PU composites and only tensile properties have been studied. Wang et al.19 reported the mechanical properties of in situ polymerized PU with different wt% of graphene nanosheets (GNS) and showed that the tensile strength and Young's modulus increased up to 239% and 202%, respectively with 2.0 wt% incorporation of GNS. Kim et al.20 reported a 10 fold increase in tensile stiffness of thermally reduced graphene (TRG)–PU composites with 3.0 wt% loading of TRG. Wu et al.21 showed that by incorporating 2.5 wt% hyper-branched aromatic polyamide functionalized graphene sheets (HBA-GNS) in PU matrix the tensile strength got enhanced by 105%. Moreover, these studies do not discuss the nano-mechanical properties of these composites. Therefore, to the best of our awareness, only one study by Cai et al.22 reported the nano-mechanical properties but discussed only two parameters such as hardness and elastic modulus but other several parameters which are important to determine the super hard behavior, elastic plastic deformation, elastic recovery etc., did not discussed in their study.
Present work describes the synthesis of RGO, fabrication of composites using PU as a polymer matrix and RGO as reinforcing material to study the nano-mechanical properties of RGO–polyurethane composites. The effects of different weight % of RGO loading on the nano-mechanical parameters of the composites are also explored. The nano-mechanical parameters estimated in the present study are; hardness (H), elastic modulus (E), plastic index parameter (H/E), elastic recovery (ER), load displacement curve, ratio between residual displacement after load removal and displacement at maximum load (hres/hmax) and plastic deformation energy. These nano-mechanical properties were also correlated with the results of SEM and Raman studies.
2. Experimental
2.1. Materials
The pure polyurethane resin thermoplastic LPR5725EG grade was purchased from C.O.I.M. LARIPUR with specific gravity of 1.23 g cm−3 and softening point of 138 °C.2.2. Synthesis of graphene oxide (GO) and reduced graphene oxide (RGO)
In the present study, improved Hummer's method23 was used for the oxidation of natural graphite (99.9% pure, -500 mesh powder) in the mixture of conc. H2SO4/H3PO4 and shown in Scheme 1 (Step 1). The reason for selecting this method was its simpler protocol and no evolution of toxic gas during preparation. The sol was stirred on a magnetic stirrer followed by slow addition of KMnO4 to avoid any explosion.24 Prior to the washing and filtration of the material with ethanol, it was ultrasonicated for 8 h in ethanol (200 ml) to exfoliate graphite oxide in to graphene oxide (GO) sheets. As prepared GO was then dried overnight in a vacuum oven at 120 °C.23 | ||
| Scheme 1 (Step 1) Process for the synthesis of graphene oxide using improved Hummer's method and (Step 2) formation of RGO reinforced PU composites for nano-mechanical properties measurements. | ||
Thermal-mediated method was used to reduce GO in which exfoliation of the stacked structure occurs through the extrusion of carbon dioxide generated by heating of GO at 1050 °C in a furnace.24 Reduction process of GO left behind vacancies and topological defects throughout the plane of the RGO platelets25,26 which are useful for better interaction with polymer matrix.
2.3. Preparation of RGO–PU composites
Composites with different RGO loadings i.e. 0.1, 0.5, 1.0, 3.0 and 5.0 wt% in PU were prepared by solvent casting technique as shown in Scheme 1 (Step 2) and hereafter will be designated as PRG0.1, PRG0.5, PRG1, PRG3 and PRG5, respectively. A pure PU sample (0% RGO) was also prepared for comparison and will be designated as PRG0. The detailed process for the synthesis of composites is already discussed in earlier study27 and given in Scheme 2 which represents the coating of PU on the surface of RGO. The coating and bonding of PU with RGO is further confirmed by TEM studies of RGO–PU composite film. | ||
| Scheme 2 Synthesis process for the formation of stretchable and flexible RGO reinforced PU composites film. | ||
2.4. Characterization
Surface morphologies of GO, RGO and fractured surface of RGO–PU composite films were investigated using SEM (Leo 440S, UK) and microstructural properties were examined using HRTEM (Technai G20-stwin, 300 kV instrument). The XRD patterns were recorded using a Bruker D8 Advance X-ray diffractometer in the diffraction (2θ) range of 10–70° (slit width of 0.1 mm) using the Cu Kα line (λ = 1.5405 Å) as radiation source.Raman studies of GO and RGO were carried out using Renishaw inVia Raman spectrometer, UK with an excitation source of 514 nm and PU and RGO–PU composite films at 785 nm. The nanoindentation study was carried out using IBIS-Nanoindentation (M/S Fisher-Cripps Laboratories Pvt. Limited, Australia), equipped with Berkovich indenter and the other details are given elsewhere.28 The analysis of the functional groups attached to the GO and RGO planes were studied by FTIR (NICOLET 5700) techniques.
In order to make the samples for TEM studies, films were grinded to make it thin (∼200 μm). Circular slice of 2.3 mm is cut using ultrasonic cutter. The slice is polished and dimple grinded to make it electron transparent (∼50 nm at centre).
3. Results and discussion
3.1. Morphological and structural studies of GO and RGO
Morphology of GO and RGO was studied using scanning electron microscope (SEM) and transmission electron microscope (TEM). Fig. 1(a) shows SEM micrograph of GO which shows carpet or sheet like morphology. Large number of wrinkles are also seen on the surface and these rough surfaces act as defects. Fig. 1(b) shows SEM micrograph of GO after reduction i.e. RGO. The micrograph clearly show high quality few layered graphene sheets which are thin, crumpled and randomly aggregated. Few single layer of graphene sheets are seen as well and the wrinkled structures also show a petal like morphology. Fig. 1(c) shows low magnification TEM micrograph in which large amount of graphene sheets are observed which are entangled with each other and some wrinkles are also seen. Fig. 1(d) shows high magnification TEM micrographs which resembles crumpled silk curtain wave like structure and some transparent regions are also seen confirming the presence of monolayer graphene. HRTEM micrograph of RGO (Fig. 1(e)) is also recorded and the magnified view of certain portion is shown in inset of Fig. 1(e) which confirms the presence of hexagons in graphene sheet. EDX analysis of RGO is carried out and the results are shown in Fig. 1(f). The elemental composition is shown in tables of the inset of Fig. 1(f). The RGO contains ∼25.83% oxygen and ∼72.77% carbon. The presence of oxygen may be due to the energetic defect sites on the edge and the basal planes of graphene sheet. It is interesting to note that RGO contains some defect sites in the form of oxygen, which is very useful for the strong interaction between RGO and organic polymer matrix. | ||
| Fig. 1 SEM micrograph of GO (a) and RGO (b), TEM micrographs of RGO at low magnification (c) and high magnification (d), HRTEM micrograph (e) and EDX spectra (f) of RGO. Raman spectra (g) and FTIR spectra (h) of GO and RGO. | ||
X-ray diffraction (XRD), Raman spectroscopy and FTIR spectroscopy were used to investigate the chemical structure, composition, presence of graphitic structures and attachment of functional groups of GO and RGO. Fig. 1(g) and (h) shows the comparison of Raman and FTIR spectra, respectively of GO and RGO.
Graphite was oxidized chemically to produce GO. The Raman spectrum of GO shows two peaks at 1597 cm−1 and 1353 cm−1 (Fig. 1(g)) which are due to the G-band and D-band, respectively. As it is a highly defective structure consisting of oxygen containing functional groups intercalated between the graphitic layers thereby reducing the π–π interaction between the layers and thus a flat 2D region (very low intensity) is observed.
The Raman spectrum of RGO (Fig. 1(g)) shows G-band and D-band at 1588 cm−1 and 1350 cm−1, respectively. The 2D band is important for the estimation of number of layers in graphene. A sharp peak at 2700 cm−1 (2D region) of intensity greater than four times of G-band intensity represents a single layer graphene29 while a modulated bump at 2D band shows few layer graphene. Thus, a significant change occurs in the shape and intensity of the 2D peak of graphene compared to bulk graphite.30
The FTIR spectrum of GO (Fig. 1(h)) shows a broad band at 3426 cm−1 in the high frequency area corresponding to the stretching vibration of –OH groups of water molecules adsorbed on graphene oxide.
XRD is important tool for both structural and compositional studies. The intensity and broadening of the peak at 2θ value of 26° corresponding to the (002) planes is very important to study the structure of a graphitic material by XRD technique.30 Oxidation of graphite causes shifting in peak position from 26° to near 10° and discussed in details in the ESI, Fig. S1 and S2.†
Therefore, it can be concluded that the sample has strong hydrophilicity, while the presence of two absorption bands observed in the medium frequency area, at 1615 cm−1 and 1720 cm−1 can be attributed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO of carboxylic acid and carbonyl groups, respectively present at the edges of graphene oxide. Finally, the absorption bands at 1148 cm−1 and 1028 cm−1 are correspond to the stretching vibration of C–OH of alcohol and C–O of C–O–C in epoxide, respectively. The presence of these oxygen containing groups reveals that the graphite has been oxidized.
C and CO of carboxylic acid and carbonyl groups, respectively present at the edges of graphene oxide. Finally, the absorption bands at 1148 cm−1 and 1028 cm−1 are correspond to the stretching vibration of C–OH of alcohol and C–O of C–O–C in epoxide, respectively. The presence of these oxygen containing groups reveals that the graphite has been oxidized.
Upon reduction of GO to RGO, the broad band of hydroxyl group remains the same, the CO band disappears and new bands at 2930 cm−1 and 2850 cm−1 arise representing the symmetric and anti-symmetric C–H stretching vibrations of the methylene group and the phenol CC ring stretching at 1578 cm−1 is present (Fig. 1(h)).
3.2. Nano-mechanical properties determined by nanoindentation
Nanoindentation properties are discussed in a previous study27 where acid modified MWCNTs was used to enhance these properties of PU composites. Herein, different wt% (0, 0.1, 0.5, 1, 3 and 5 wt%) of RGO in PU matrix has been used and very significant changes in the mechanical properties have been observed. The theory and equations related to the nanoindentation properties have already been discussed earlier.27 Mechanical properties such as hardness (H), elastic modulus (E), elastic recovery (% ER) and load displacement curves were determined by nanoindenter test. The load versus displacement curves at minimum indentation load of 1 mN for PRG0, PRG0.1, PRG0.5, PRG1, PRG3 and PRG5 composites have been studied and are shown in Fig. 2(a–f), respectively. These load displacement curves show the recovery of the composite films after load removal. Load versus displacement curves were further used to estimate the several other elastic and plastic properties of these composites. | ||
| Fig. 2 Load versus displacement curves of (a) PRG0, (b) PRG0.1, (c) PRG0.5, (d) PRG1, (e) PRG3 and (f) PRG5 at 1 mN load. | ||
It has been reported in the previous studies that indentation hardness and other related mechanical properties depend upon the substrate and nature of the film.31 In the present study, self-supporting free standing RGO–PU composite sheets have been prepared which avoid the substrate effect on the mechanical properties.
From Fig. 3, it is observed that the value of hardness increases with increase in the amount of RGO. The value of hardness for pure PU sample (PRG0) was 58.5 MPa which increased to 140 MPa for PRG5. Similarly the elastic modulus increased from 385.7 MPa for PRG0 to 881.7 MPa for PRG5. Thus, an overall improvement of 139% in the hardness and 129% in the elastic modulus was achieved with RGO reinforcement.
 | ||
| Fig. 3 Variation of hardness and elastic modulus at 1 mN indentation load. | ||
This enhancement in the hardness and elastic modulus of RGO reinforced PU composites could be due to the presence of a strong interaction and bonding between the functional groups of RGO and the PU chains.
The strong interaction between RGO and PU is confirmed by SEM studies of fractured surfaces of RGO–PU composites as shown in Fig. 4. From Fig. 4(a) and (b) for PRG0.5 and Fig. 4(c) and (d) for PRG1 at different magnifications, it can be seen that RGO remains properly embedded in PU matrix after fracture because large amount of functional groups are attached on the edge and basal planes of RGO which makes strong hydrogen bonding with PU chains. The increase in hardness of the polymer composites with increase in the amount of RGO is due to strong interaction between them. In order to further investigate the interaction and bonding between RGO and PU composites; Raman spectroscopic analysis of RGO, PU and RGO–PU composites has been carried out. The results are interpreted based on changes in Raman shift (as shown in Fig. 5). Raman shift can provide insight into dispersion and interaction of RGO in PU matrix.10,32,33
 | ||
| Fig. 4 SEM micrograph of the fractured surface of (a and b) PRG0.5 and (c and d) PRG1. | ||
 | ||
| Fig. 5 Raman spectra of (a) PRG0, (b) PRG0.1, PRG0.5, PRG1, PRG3 and PRG5, (c) magnified view of Raman shift and (d) variation in Raman shift with RGO loading of different composites. | ||
Fig. 5(a) shows the Raman spectrum of PRG0 (pure PU). The peak positions are already discussed in previous study.27 After addition of RGO, the intensity of Raman peaks of pure PU starts decreasing and finally disappears for higher loading in the case of PRG3 and PRG5 (Fig. 5(b)), which shows that RGO dominates over PU. The shifting of the Raman peak of carbonyl group of PU towards left side i.e. lower value in RGO–PU composites shows interaction between RGO and PU chain.
Fig. 5(c) shows magnified view of the shifting in Raman peak of RGO–PU composites and the changes in the Raman shift is shown in Fig. 5(d). The band at 1611 cm−1 for carbonyl group of pure PU shifts from 1611–1609 cm−1 in PRG0.1 to 1611–1607 cm−1 in PRG0.5 and 1611–1603 cm−1 in PRG1. The total shift of 8 cm−1 in the Raman peak shows the good dispersion and interaction between functional groups of RGO and PU chain.27 It was not possible to observe the peak position of PU in PRG3 and PRG5 samples because of dominance of RGO in the Raman spectrum.
Fig. 6 shows the TEM micrographs of 1 wt% RGO–PU composite films. Fig. 6(a) shows that RGO is dispersed uniformly in the PU matrix and are surrounded by the PU matrix (shown by yellow dashed circles). Fig. 6(b) shows that the RGO is encapsulated in the polymer matrix and there is a strong interaction at the surface of the RGO and polymer because RGO appeared thickly coated. Presence of graphene planes is clearly seen and marked in Fig. 6(b).
 | ||
| Fig. 6 TEM micrograph of (a) RGO embedded in the PU matrix for PRG1 and (b) coating of PU on RGO plane is clearly seen. | ||
In addition to hardness and elastic modulus, the wear resistant is also a very important parameter in determining the quality of the composites. Plastic resistance parameter (H/E) explains not only the elastic–plastic properties but also gives the information about wear resistance.
The variation of H/E at 1 mN load for various RGO–PU composites is shown in Fig. 7. The value of H/E is 0.158 for PRG5 and is 0.151 for pure PU (PRG0). The higher value of H/E in PRG5 reveals that the material can be useful for wear resistant coatings because hardness also increases with increase in elastic modulus.
 | ||
| Fig. 7 Plastic resistance parameter (H/E) at 1 mN indentation load. | ||
Load versus displacement curves are used to calculate the elastic recovery (ER). Since, there is a competition between elastic and plastic deformation under the load, the values of ER varies in the range of 58.0% to 67.5% for different composites (as shown in Fig. 8(a)). Soft polymer composites show plastic deformation to some extent due to surface penetration by sharp indenter. There is a slight difference in the ER values of PRG0 and PRG5 and polymer composites recover after the load removal. The hres/hmax curves for various composites are shown in Fig. 8(b). The behavior is similar to that of ER. The value of this parameter should vary between 0 and 1 in which lower limit corresponds to the fully elastic behavior and upper limit corresponds to rigid plastic behavior. Results in the present investigation vary between 0.32 and 0.42 which indicates fully elastic behavior of these composites at 1 mN load. These observed results are in good agreement with ER results.
 | ||
| Fig. 8 Variation of (a) % elastic recovery (% ER) and (b) hres/hmax with RGO loading at 1 mN indentation load. | ||
Contact stiffness (dP/dh)max is related to elastic and elastic–plastic properties of RGO–PU composites. Stiffness is defined as resistance offered by elastic body to deform under load. The variation of contact stiffness for different composites is shown in Fig. 9(a). It is evident from the figure that the composite PRG5 exhibits maximum stiffness. The value of stiffness for PRG0 is 1.38 × 103 N m−1 which increases to 2.75 × 103 N m−1 for PRG5. Thus, an overall improvement of ∼99% in stiffness is observed. Observed value of stiffness is in good agreement with the results of hardness and elastic modulus.
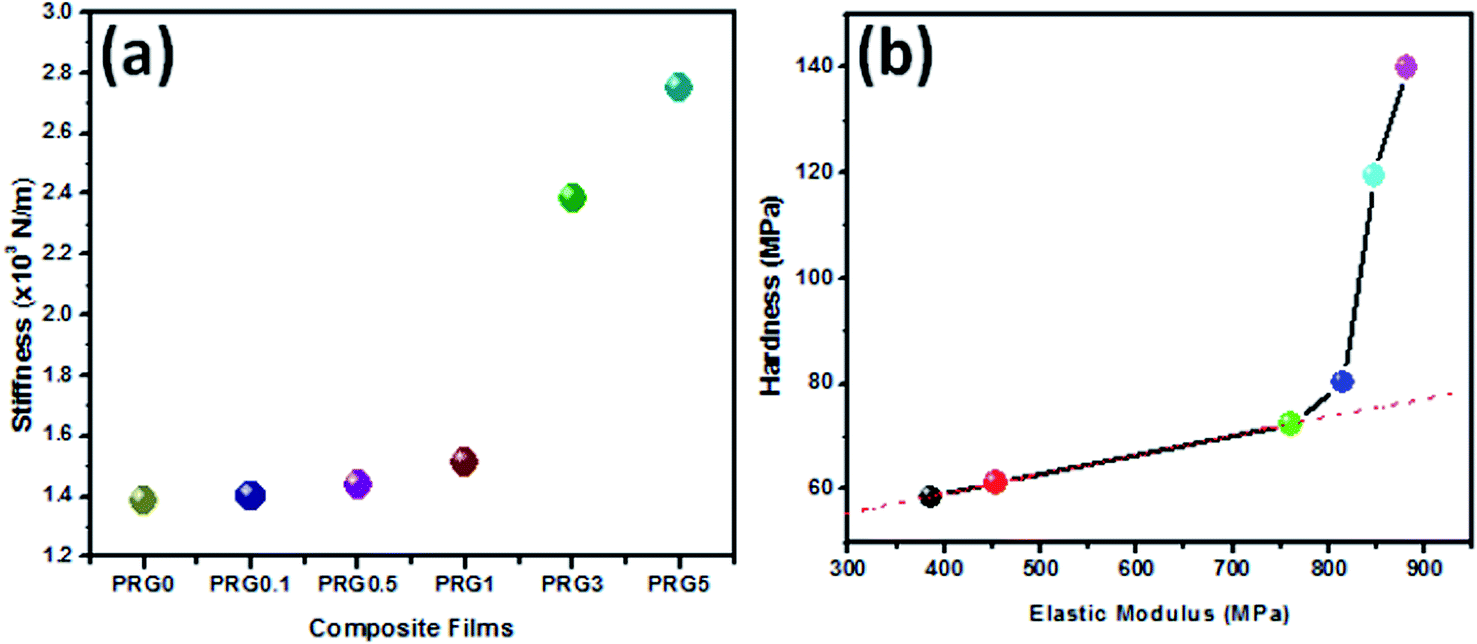 | ||
| Fig. 9 (a) Variation of stiffness with RGO loading and (b) linear behavior of H–E curve for different type of RGO–PU composites. | ||
Hardness (H) strongly depends on the presence of nano to microstructural defect in the network, and it is related to the bonding between the atoms and to the ability of the bonds to withstand deformation stemming from compression, extension, bending, or breaking, whereas E depends on the slope of harmonic interatomic potential. The H versus E behavior of RGO–PU composites is shown in Fig. 9(b). Generally, H varies linearly with respect to E and follow the relation H = (1/10)E. As seen in the beginning follow linear path, but for increased loading of RGO their linear path deviates and shows H ≥ (1/10)E behavior. Thus, one can see that instead of the conventional linear behavior (represented by red dashed line in the Fig. 9(b)), these composites exhibit super-linear behavior (represented by dark line) after certain loading of RGO, which is due to increased toughness of the structures.
The elastic and plastic properties of these RGO–PU composites in terms of plastic deformation energy (Ur) have also been calculated. The theory and equation of deformation energy is already discussed.27,31
The variation of Ur for different loading of RGO in PU composites is shown in Fig. 10. It is to be noted that Ur is inversely proportional to the hardness (H).27 Ur results follow the said statement. The minimum value of Ur is 3.61 × 10−10 J at 1 mN load for PRG5. The results of Ur are also in good agreement with the results of Hardness.
 | ||
| Fig. 10 Deformation energy (Ur) of different RGO–PU composites. | ||
These RGO–PU composites can also be used as shape memory polymer composites as polyurethane has been previously used by several researchers.34–39
4. Conclusion
RGO–PU composites have been fabricated by solvent casting technique and significant enhancement in mechanical properties is observed. An overall enhancement of 139% in the hardness and 129% in elastic modulus were observed in nanoindentation tests. Addition of RGO into the PU matrix not only reduced the deformation energy considerably but also improved the stiffness and other mechanical parameters significantly. The fractured surface of these composites (SEM) clearly revealed the strong interaction of RGO with PU chain. This strong interaction was also confirmed by Raman studies of RGO–PU composites which revealed by the significant shifting in Raman peaks. The strong interaction between RGO and PU and the presence of graphene planes were also confirmed by TEM studies of RGO–PU composite films. These composites can find industrial applications as hard and scratchless coating in automobile industry.Acknowledgements
The authors are grateful to Prof. R. C. Budhani, Director, CSIR-National Physical Laboratory, New Delhi, India for his kind support and encouragement. The authors also wish to thank Mr K. N. Sood and Mr Jai Tawale for SEM characterization and Mr Dinesh Singh for the preparation of the sample for TEM measurements. The studies were carried out under CSIR network project (PSC0109). One of the authors (TKG) acknowledges CSIR, Govt. of India, for providing a senior research fellowship (SRF).References
- D. D. Evanoff and G. Chumanov, ChemPhysChem, 2005, 6, 1221–1231 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. B. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- D. Y. Godovsky, in Biopolymers PVA Hydrogels, Anionic Polymerisation Nanocomposites, Springer, 2000, pp. 163–205 Search PubMed.
- M. S. Dresselhaus and G. Dresselhaus, Adv. Phys., 1981, 30, 139–326 CrossRef CAS.
- M. Hirata, T. Gotou, S. Horiuchi, M. Fujiwara and M. Ohba, Carbon, 2004, 42, 2929–2937 CAS.
- M.-F. Yu, O. Lourie, M. J. Dyer, K. Moloni, T. F. Kelly and R. S. Ruoff, Science, 2000, 287, 637–640 CrossRef CAS.
- H. Tobushi, S. Hayashi, K. Hoshio and Y. Ejiri, Sci. Technol. Adv. Mater., 2008, 9, 015009 CrossRef.
- C. Hepburn, Polyurethane elastomers, Elsevier, Essex, 1992, 2nd edn, p. 1 Search PubMed.
- S. Gogolewski, Rev. Colloid Polym. Sci., 1989, 267, 757–785 CrossRef CAS.
- S. M. Xia, Soft Mater, 2005, 1, 386–394 RSC.
- C. Hepburn, Polyurethane elastomers, Applied Science Publishers, London, 1982 Search PubMed.
- S. Husić, I. Javni and Z. S. Petrović, Compos. Sci. Technol., 2005, 65, 19–25 CrossRef PubMed.
- W. D. Zhang, L. Shen, I. Y. Phang and T. Liu, Macromolecules, 2004, 37, 256–259 CrossRef CAS.
- J. N. Coleman, M. Cadek, R. Blake, V. Nicolosi, K. P. Ryan, C. Belton, A. Fonseca, J. B. Nagy, Y. K. Gun'ko and W. J. Blau, Adv. Funct. Mater., 2004, 14, 791–798 CrossRef CAS.
- J. C. Grunlan, A. R. Mehrabi, M. V. Bannon and J. L. Bahr, Adv. Mater., 2004, 16, 150–153 CrossRef CAS.
- H. Huang, C. Liu, Y. Wu and S. Fan, Adv. Mater., 2005, 17, 1652–1656 CrossRef CAS.
- M. Muralidharan and S. Ansari, Adv. Mater. Lett., 2013, 4, 927–932 CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- X. Wang, Y. Hu, L. Song, H. Yang, W. Xing and H. Lu, J. Mater. Chem., 2011, 21, 4222–4227 RSC.
- H. Kim, Y. Miura and C. W. Macosko, Chem. Mater., 2010, 22, 3441–3450 CrossRef CAS.
- C. Wu, X. Huang, G. Wang, X. Wu, K. Yang, S. Li and P. Jiang, J. Mater. Chem., 2012, 22, 7010–7019 RSC.
- D. Cai, K. Yusoh and M. Song, Nanotechnology, 2009, 20, 085712 CrossRef PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- T. Nakajima, A. Mabuchi and R. Hagiwara, Carbon, 1988, 26, 357–361 CrossRef CAS.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS PubMed.
- T. K. Gupta, B. P. Singh, S. R. Dhakate, V. N. Singh and R. B. Mathur, J. Mater. Chem. A, 2013, 1, 9138–9149 CAS.
- S. Kumar, N. Dwivedi, C. M. S. Rauthan and O. S. Panwar, Appl. Phys. A: Mater. Sci. Process., 2011, 102, 225–230 CrossRef.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- R. Kamaliya, B. P. Singh, B. K. Gupta, V. N. Singh, T. K. Gupta, R. Gupta, P. Kumar and R. B. Mathur, Carbon, 2014, 78, 147–155 CrossRef CAS PubMed.
- S. Kumar, N. Dwivedi and H. K. Malik, Appl. Surf. Sci., 2011, 257, 9953–9959 CrossRef PubMed.
- M. Lucas and R. J. Young, Compos. Sci. Technol., 2004, 64, 2297–2302 CrossRef CAS PubMed.
- D. A. Heller, P. W. Barone, J. P. Swanson, R. M. Mayrhofer and M. S. Strano, J. Phys. Chem. B, 2004, 108, 6905–6909 CrossRef CAS.
- M. Momtaz, M. Razavi-Nouri and M. Barikani, J. Mater. Sci., 2014, 49, 7575–7584 CrossRef CAS.
- S. Weng, Z. Xia, J. Chen and L. Gong, J. Appl. Polym. Sci., 2013, 127, 748–759 CrossRef.
- M. Bothe, F. Emmerling and T. Pretsch, Macromol. Chem. Phys., 2013, 214, 2683–2693 CrossRef CAS.
- K. Hearon, K. Gall, T. Ware, D. J. Maitland, J. P. Bearinger and T. S. Wilson, J. Appl. Polym. Sci., 2011, 121, 144–153 CrossRef CAS PubMed.
- M. Ecker and T. Pretsch, RSC Adv., 2014, 4, 46680–46688 RSC.
- M. Ecker and T. Pretsch, RSC Adv., 2014, 4, 286–292 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra14223c |
| This journal is © The Royal Society of Chemistry 2015 |