DOI:
10.1039/C4RA14122A
(Paper)
RSC Adv., 2015,
5, 12442-12445
Domino reaction of N-(cyanomethyl)-1,3-azolium quaternary salts with o-hydroxybenzaldehydes: scope and limitations†
Received
8th November 2014
, Accepted 13th January 2015
First published on 14th January 2015
Abstract
A route towards chromenes, annulated with an imidazo[5,1-c][1,4]thiazine core through a base-promoted domino reaction of thiazolium quaternary salts, has been developed. The synthesised compounds show high cytotoxic activity against human tumour cell lines.
Introduction
Domino reactions, also known as tandem or cascade reactions, have emerged as a highly effective strategy for the synthesis of heterocyclic compounds, including bioactive natural products and pharmaceutical agents.1 These protocols enable chemists to perform complex synthetic conversions with high efficiency using readily available starting materials, often via a biomimetic pathway.2 Thus, domino reactions contribute exceedingly to synthetic drug design strategies, enhance elegant approaches in total synthesis and improve yields in large-scale syntheses.1,2 The advantages of these methods include excellent atom economy, high selectivity and less waste.3 Additionally, using these strategies, multiple transformations can be carried out in a single laboratory operation without the isolation of intermediates, making them prime examples of green chemistry.4 Despite the widespread proliferation of domino reactions, researchers have continued to channel their efforts in this area, as new heterocyclic structures and novel substitution patterns are required.5
The reactivity of N-(cyanomethyl) heterocyclic quaternary salts in domino reactions are of interest, owing to the structural complexity generated and the potential biological activity of the resulting products. Investigations in this field have shown the possibility to easily transform pyridinium salts to chromenoimidazopyridines,6 isoquinolinium salts to chromeno-7 and thiochromeno-imidazoisoquinolines.8 Moreover, we have had preliminary results showing a route to the chromenoimidazothiazine core through the ANRORC transformation of N-(cyanomethyl)-1,3-thiazolium salts under the action of salicylic aldehydes.9 The optimisation of the latter reaction conditions, the extension of the methodology to other 1,3-azoles and the biological evaluation of the chromeno-imidazothiazines are disclosed in the present paper.
Results and discussion
Synthesis of N-(cyanomethyl)-1,3-azolium salts
The preparation of the starting thiazolium salts, 1a–c, has previously been reported.9 The yields can be significantly increased by running the reactions under microwave (MW) irradiation conditions (Table 1). Imidazolium salt 2 was prepared with a good yield without employing MW irradiation. Unfortunately, we did not succeed in preparing the oxazolium quaternary salts by any means; the use of more facile leaving groups (–Br, –I), solvent-free techniques and MW irradiation did not provide positive results.
Table 1 The synthesis of quaternary salts 1, 2

|
| Product |
R1 |
R2 |
X |
Conditions |
Prev. rep. yield, % |
Yield, % |
| 1a |
H |
H |
S |
MW, 140 °C, 30 min, solvent-free |
49 |
81 |
| 1b |
Me |
H |
S |
MW, 140 °C, 30 min, solvent-free |
33 |
79 |
| 1c |
Me |
Me |
S |
MW, 140 °C, 30 min, solvent-free |
20 |
82 |
| 1d |
H |
Me |
S |
MW, 140 °C, 30 min, solvent-free |
65 |
81 |
| 2 |
H |
H |
N–Me |
CH3CN, 50 °C, 1 h |
— |
78 |
Reaction of thiazolium salts with o-hydroxybenzaldehydes: optimisation and scope
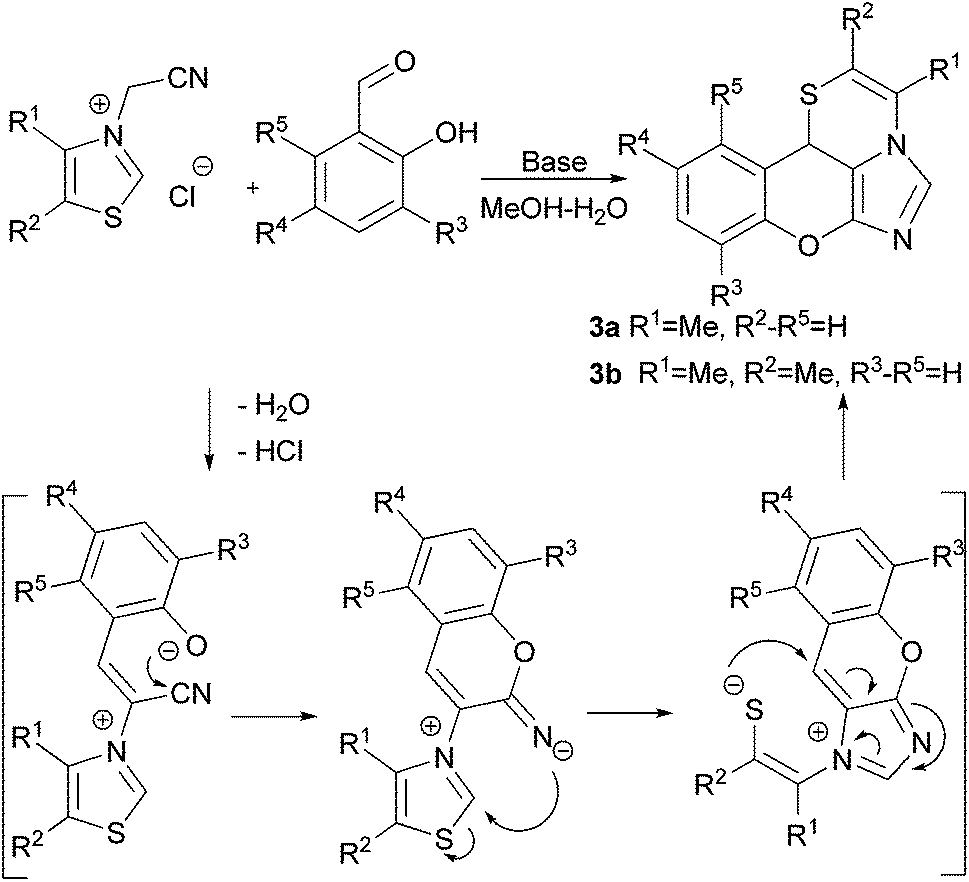
Owing to preliminary studies,9 the reaction of thiazolium salts 1 with o-hydroxybenzaldehydes under base-promoted conditions proceeded as a domino process, involving an ANRORC step, and led to the formation of chromenoimidazothiazines 3 (Scheme 1).
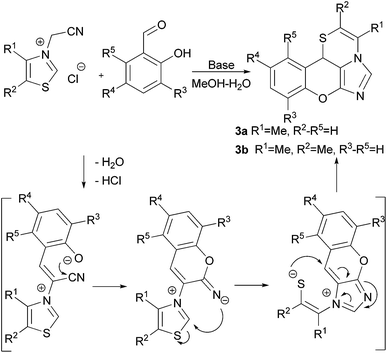 |
| | Scheme 1 Plausible scheme of chromenoimidazothiazine formation. | |
The initial optimisation of the reaction conditions showed that the use of 20 mol% sodium carbonate as a base and MeOH–H2O as a solvent was optimal. Still, the yields of the tetracyclic products were satisfactory, but the reaction failed to produce target compounds with salicylic aldehyde (R3, R4, R5 = H). To overcome these problems, a more thorough study of the reaction conditions was initiated. The reactions of thiazolium salts 1b and 1c with salicylic aldehyde were chosen as the model, and the results of the optimisation process are summarised in Table 2. It has been shown that the use of promoters such as ammonium acetate, potassium tert-butoxide, L-proline, triethylamine or dimethylaminopyridine resulted in the formation of only trace amounts of products 3a and 3b (Table 2, entries 1–6). The use of potassium carbonate (20 mol%) provided compound 3b in 18% yield in refluxing MeOH–H2O for 3 h. The use of 60 mol% K2CO3 raised the yield to 38% with 3 h reflux. A further increase in the amount of K2CO3 (100 mol%) provided compound 3b in 48% yield after 10 min reflux, but resulted in complex-mixture formation in the case of 3a (Table 2, entries 10 and 11). The employment of TFE or DMF as solvents did not result in any yield improvements (Table 2, entries 13–15). DBU was found to be the most suitable base, as compound 3b was obtained in 61% yield and 3a in 62% yield. Further studies failed to improve these yields. The methanol and water were not used separately due to the poor solubility of the quaternary salts in pure alcohol and substituted aldehydes in pure water. As far as the products of the reactions precipitate from the reaction mixture, the homogeneity of the starting reactants in the solvent is important for producing the precipitates with the acceptable purity. The general recommendations for carrying out these reactions are the avoidance of high temperatures and to minimise the reaction time when using either an equivalent or excess amount of base.
Table 2 Optimisation of the model reaction conditions
| Entry |
T, °C |
t, h |
Solvent |
Promoter |
Prod. |
Yield, % |
| 1 |
Reflux |
1 |
MeOH–H2O |
NH4OAc (100 mol%) |
3b |
10 |
| 2 |
Reflux |
3 |
MeOH–H2O–THF |
t-BuOK (20 mol%) |
3a |
Trace |
| 3 |
r.t. |
12 |
MeOH–H2O |
L-Proline (10 mol%) |
3a |
Trace |
| 4 |
Reflux |
3 |
MeOH–H2O |
L-Proline (120 mol%) |
3a |
Trace |
| 5 |
r.t. |
3 |
MeOH–H2O |
Et3N (100 mol%) |
3a |
7 |
| 6 |
Reflux |
3 |
MeOH–H2O |
DMAP (100 mol%) |
3a |
Trace |
| 7 |
Reflux |
3 |
MeOH–H2O |
K2CO3 (20 mol%) |
3b |
18 |
| 8 |
Reflux |
1 |
MeOH–H2O |
K2CO3 (20 mol%) |
3a |
19 |
| 9 |
Reflux |
3 |
MeOH–H2O |
K2CO3 (60 mol%) |
3b |
38 |
| 10 |
Reflux |
0.1 |
MeOH–H2O |
K2CO3 (100 mol%) |
3b |
48 |
| 11 |
Reflux |
0.1 |
MeOH–H2O |
K2CO3 (100 mol%) |
3a |
Trace |
| 12 |
40 |
1 |
MeOH–H2O |
K2CO3 (100 mol%) |
3a |
37 |
| 13 |
80 |
1 |
DMF |
K2CO3 (20 mol%) |
3a |
Trace |
| 14 |
80 |
1 |
DMF |
K2CO3 (100 mol%) |
3a |
Trace |
| 15 |
Reflux |
0.1 |
TFE |
K2CO3 (100 mol%) |
3a |
10 |
| 16 |
Reflux |
1 |
MeOH–H2O |
DBU (100 mol%) |
3b |
43 |
| 17 |
r.t. |
18 |
MeOH–H2O |
DBU (110 mol%) |
3b |
61 |
| 18 |
r.t |
18 |
MeOH–H2O |
DBU (110 mol%) |
3a |
62 |
To show the advantages of the newly selected conditions, previously reported compounds were obtained by a modified protocol. Thus, the yields of compounds 3d, 3e, 3g and 3h were significantly improved (Table 3, entries 4–7 and 9–12). The reaction worked well for aldehydes bearing both electron-donating and electron-withdrawing groups, giving target compounds 3 with satisfactory-to-good yields (Table 3).
Table 3 The scope of chromenoimidazothiazines 3
| Entry |
R1 |
R2 |
R3 |
R4 |
R5 |
Conditions |
Product |
Yield, % |
| 1 |
Me |
H |
H |
H |
H |
DBU (110 mol%), r.t., 18 h |
3a |
62 |
| 2 |
Me |
Me |
H |
H |
H |
DBU (110 mol%), r.t., 18 h |
3b |
61 |
| 3 |
H |
Me |
H |
H |
H |
K2CO3 (110 mol%), 40 °C, 1 h |
3c |
34 |
| 4 |
Me |
H |
H |
Br |
H |
Na2CO3 (20 mol%), reflux, 1 h |
3d |
28 |
| 5 |
Me |
H |
H |
Br |
H |
DBU (110 mol%), r.t., 18 h |
3d |
61 |
| 6 |
Me |
Me |
H |
Br |
H |
Na2CO3 (20 mol%), reflux, 1 h |
3e |
12 |
| 7 |
Me |
Me |
H |
Br |
H |
DBU (110 mol%), r.t., 18 h |
3e |
61 |
| 8 |
H |
Me |
H |
Br |
H |
DBU (110 mol%), r.t., 12 h |
3f |
46 |
| 9 |
Me |
H |
H |
NO2 |
H |
Na2CO3 (20 mol%), reflux, 1 h |
3g |
27 |
| 10 |
Me |
H |
H |
NO2 |
H |
DBU (110 mol%), r.t., 18 h |
3g |
81 |
| 11 |
Me |
Me |
H |
NO2 |
H |
Na2CO3 (20 mol%), reflux, 1 h |
3h |
12 |
| 12 |
Me |
Me |
H |
NO2 |
H |
DBU (110 mol%), r.t., 18 h |
3h |
76 |
| 13 |
H |
Me |
H |
NO2 |
H |
DBU (110 mol%), r.t., 12 h |
3i |
34 |
| 14 |
Me |
Me |
H |
–CH–(CH)2– |
CH– |
K2CO3 (20 mol%), reflux, 45 min |
3j |
30 |
| 15 |
H |
Me |
H |
–CH–(CH)2– |
CH– |
K2CO3 (20 mol%), reflux, 45 min |
3k |
37 |
| 16 |
Me |
H |
H |
OMe |
H |
K2CO3 (110 mol%), 40 °C, 1 h |
3l |
54 |
| 17 |
Me |
Me |
H |
OMe |
H |
K2CO3 (110 mol%), 40 °C, 1 h |
3m |
72 |
| 18 |
H |
Me |
H |
OMe |
H |
K2CO3 (110 mol%), 40 °C, 1 h |
3n |
58 |
| 19 |
Me |
Me |
OMe |
NO2 |
H |
DBU (110 mol%), r.t., 18 h |
3o |
43 |
Reactions of imidazolium salt with o-hydroxybenzaldehydes
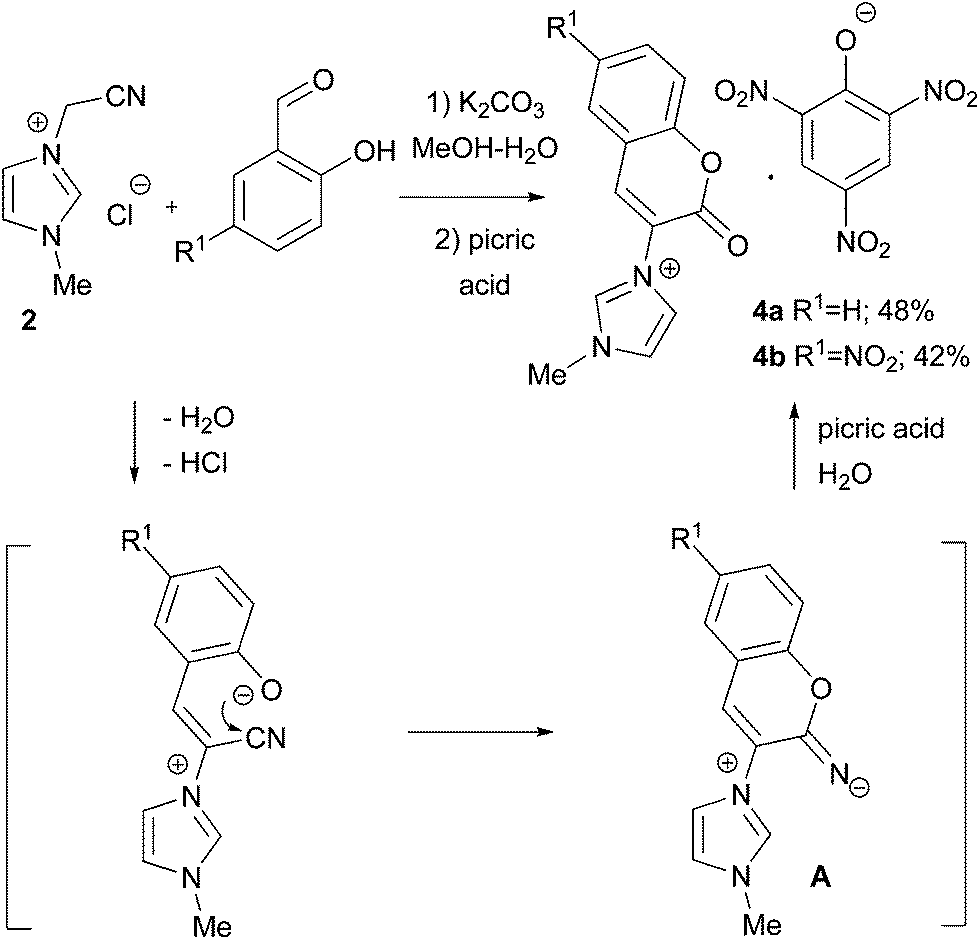
The reactions of imidazolium salt 2 with o-hydroxybenzaldehydes were anticipated to proceed in a similar way. Despite the expectations, the reaction of 2 and salicylic aldehyde in MeOH–H2O, using K2CO3 as a base, produced no mobile spots on the TLC plate. The resulting product 4a precipitated from the reaction mixture after the addition of picric acid, making it possible to characterise the products by X-ray analysis.10 The formation of this coumaryl-substituted imidazolium picrate may be explained by the hydrolysis of the 42% yield. To avoid hydrolysis, the reaction was carried out in water-free conditions, but the exploitation of dry DMF or MeOH led to the formation of inseparable mixtures.
Biological evaluation of chromenoimidazothiazines
Compounds 3b, 3e, 3l, 3m and 5 (ref. 9) were evaluated in vitro for their cytotoxic activity against four human tumour cell lines (KB, Hep-G2, LU and MCF-7), and the results are summarised in Table 4. These particular compounds have been selected due to their better water solubility. Four chromeno-imidazothiazine derivatives showed strong activity against the KB cell line with an IC50 value below 100 μg mL−1. Analogues 3m and 3l imine bond in zwitter-ion A (Scheme 2). The similar product, 4b, was obtained from nitro-substituted salicylic aldehyde with exhibited potent cytotoxicity against the KB cell line with IC50 = 4 and 6.32 μg mL−1, respectively. Meanwhile, analogues 3e and 3l inhibited the Hep-G2 cell line with IC50 values in the 80–117.5 μg mL−1 range. Derivative 3m displayed cytotoxic activity against LU cell lines, with an IC50 value of 99.76 μg mL−1. Concerning the last cell line, MCF7, the chromenoimidazothiazines analogues showed weak activities, with IC50 values above 128 μg mL−1. It is noteworthy to mention that two derivatives, 3m and 3l, present a cytotoxicity activity against the cancer cell line KB that is comparable with ellipticine.
Table 4 Cytotoxic activity of compounds 3b, 3e, 3l, 3m and 5
| Compound |
Cell line, IC50 μg mL−1 |
| KB |
HepG2 |
Lu |
MCF-7 |
| Ellipticin |
0.25 |
0.29 |
1.18 |
0.71 |
| 3b |
32 |
>128 |
>128 |
>128 |
| 3e |
68.0 |
117.5 |
>128 |
>128 |
| 3l |
4 |
80 |
>128 |
>128 |
| 3m |
6.32 |
>128 |
99.76 |
>128 |
 |
>128 |
>128 |
>128 |
>128 |
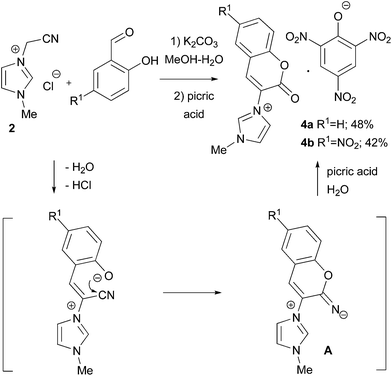 |
| | Scheme 2 Coumaryl-substituted imidazolium picrate 4 formation. | |
Conclusions
A number of 10bH-6-oxa-1-thia-3a,5-diazaacephenanthrylenes have been synthesised through the ANRORC domino reaction of N-(cyanomethyl)-1,3-thiazolium salts with salicylic aldehydes. It has been shown that 3-(cyanomethyl)-1-methylimidazolium chloride reacts with salicylic aldehydes differently to expected, forming coumaryl-substituted imidazolium salts. It has been also reported that the 1,3-oxazole failed to give the N-cyanomethyl quaternary salt. Some of the synthesised compounds were tested in vitro and showed high cytotoxic activity against human tumour cells.
Acknowledgements
This work was financially supported by the Russian Foundation for Basic Research (grants 14-03-31140-mol_a and 14-03-93001), the Ministry of education and science of Russian Federation (project 2042) and VAST (grant VAST.HTQT.NGA.06/14-15).
Notes and references
- K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef CAS PubMed.
- K. C. Nicolaou, T. Montagnon and S. A. Snyder, Chem. Commun., 2003, 551 RSC.
- L. F. Tietze, G. Brasche and K. Gericke, Domino Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 2006, pp. 160–185 Search PubMed.
- N. J. Parmar, H. A. Barad, B. M. Labana, R. Kant and V. K. Gupta, RSC Adv., 2013, 3, 20719 RSC.
- L. G. Voskressensky, A. A. Festa and A. V. Varlamov, Tetrahedron, 2014, 70, 551 CrossRef CAS PubMed.
- M. F. Proença and M. Costa, Tetrahedron, 2010, 66, 4542 CrossRef PubMed.
- L. G. Voskressensky, A. A. Festa, E. A. Sokolova and A. V. Varlamov, Tetrahedron, 2012, 68, 5498 CrossRef CAS PubMed.
- L. G. Voskressensky, E. A. Sokolova, A. A. Festa and A. V. Varlamov, Tetrahedron Lett., 2013, 54, 5172 CrossRef CAS PubMed.
- L. G. Voskressensky, A. A. Festa, E. A. Sokolova, V. N. Khrustalev and A. V. Varlamov, Eur. J. Org. Chem., 2012, 2012, 6124 CrossRef CAS.
- N. V. Tuyen, A. T. Le, A. A. Festa, L. G. Voskressensky and V. N. Khrustalev, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o839 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, copies of 1H and 13C spectra. See DOI: 10.1039/c4ra14122a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.