
A short and highly convergent approach for the synthesis of rutaecarpine derivatives†
B. V. Subba Reddy*a,
R. Anji Babuae,
B. Jagan Mohan Reddye,
B. Sridharb,
T. ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ramalinga Murthyc,
P. Pranathic,
Shasi V. Kalivendic and
T. Prabhakar Raod
Ramalinga Murthyc,
P. Pranathic,
Shasi V. Kalivendic and
T. Prabhakar Raod
aNatural Product Chemistry, CSIR-Indian Institute of Chemical Technology, Tarnaka, 500 007, Hyderabad, India. E-mail: basireddy@iict.res.in; Web: http://www.iictindia.org Fax: +91-40-27160512
bLaboratory of X-ray Crystallography, CSIR-Indian Institute of Chemical Technology, Tarnaka, 500 007, Hyderabad, India
cCentre for Chemical Biology, CSIR-Indian Institute of Chemical Technology, Tarnaka, 500 007, Hyderabad, India
dCentre for Nuclear Magnetic Resonance, CSIR-Indian Institute of Chemical Technology, Tarnaka, 500 007, Hyderabad, India
eDepartment of Organic Chemistry, Adikavi Nannaya University, Rajahmundry, 533105, India
First published on 12th March 2015
Abstract
A novel and highly convergent approach has been developed for the synthesis of quinazolinocarboline alkaloid, rutaecarpine, analogues by the condensation of anthranilamide with chloroaldehyde derived from tryptophol. This method also provides direct access to benzimidazocarbolines and quinazolinoisoquinolines. It is a shorter and metal-free approach which does not require any catalyst or additive to facilitate the reaction.
Introduction
Rutaecarpine, a naturally occurring quinazolinocarboline alkaloid isolated from the fruits of Evodia rutaecarpa1 is known to possess a diverse range of pharmacological properties such as anticancer, anti-inflammatory and anti-obesity behaviour (Fig. 1).2,3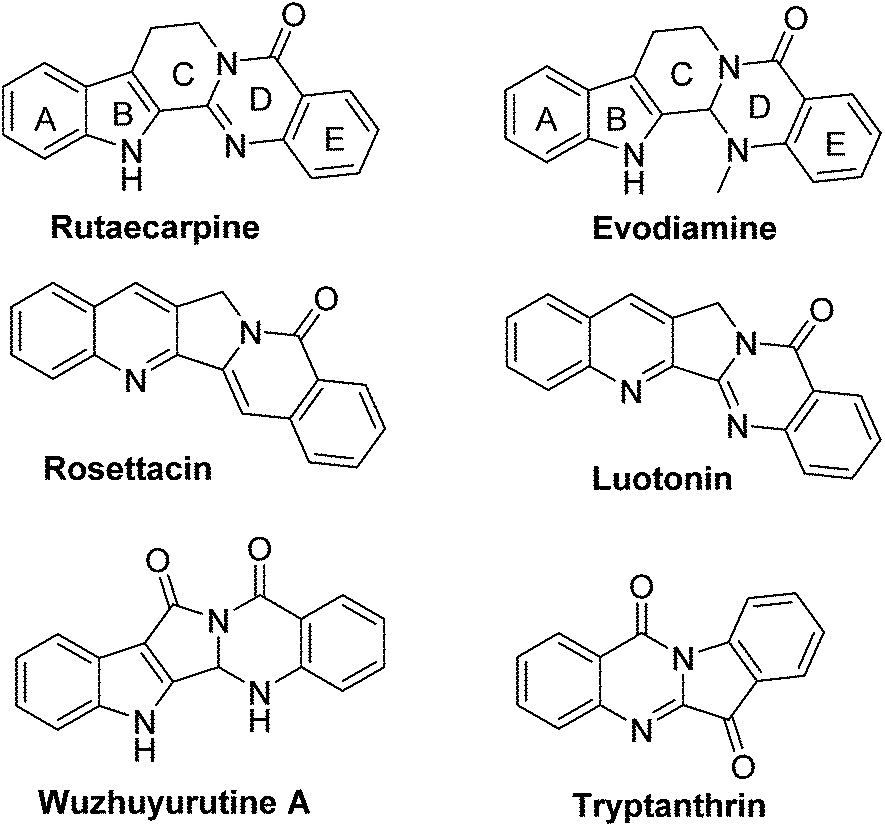 | ||
| Fig. 1 Biologically active natural products. | ||
It has also been used as a hypertensive, antiallergic, diuretic and platelet aggregation inhibitor.4 Indeed, analogues of rutaecarpine are found to display superior activity than rutaecarpine when evaluated against several tumour cell lines.5–7 Generally, rutaecarpine derivatives were synthesized by structurally modifying the A or D or E rings.8 Therefore, the development of novel approaches to access rutaecarpine analogues may lead to therapeutic applications. Consequently, several methods have been reported for the synthesis of various analogues of rutaecarpine.9 However, many of them involve multi-step synthetic sequences, stringent conditions, and expensive reagents.
Following our interest on quinazolinone alkaloids,10 we herein report a highly convergent approach for the synthesis of rutaecarpine analogues through the condensation of anthranilamide with a bifunctional chloroaldehyde. The condensation proceeds smoothly in DMF through haloamide cyclization (Scheme 1).
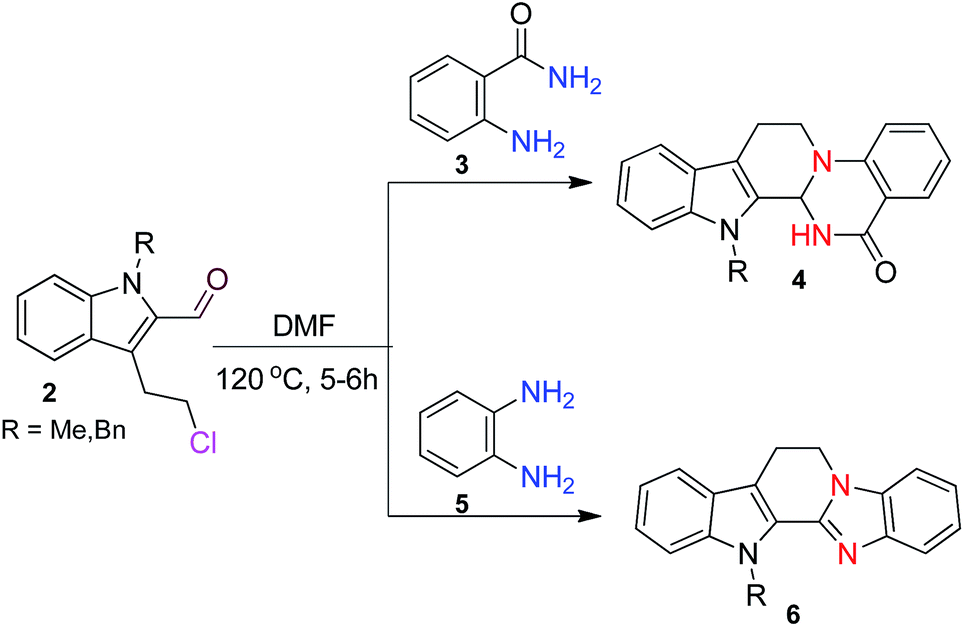 | ||
| Scheme 1 Synthesis of quinazolinocarbolines and benzimidazocarbolines. | ||
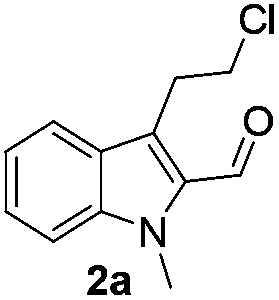
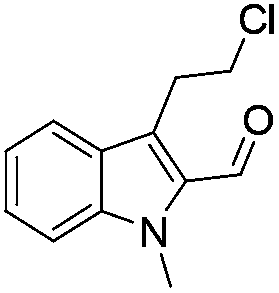
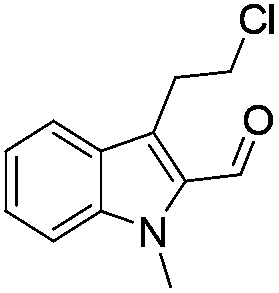
The requisite chloroaldehyde was prepared from the tryptophol through a sequential N-protection and Vilsmeier–Haack reaction (Scheme 2).
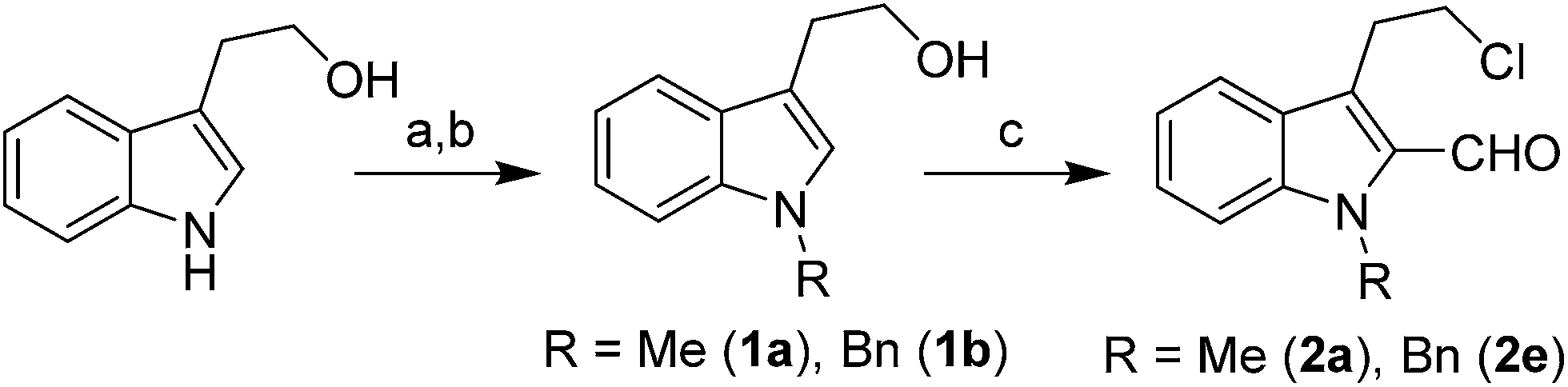 | ||
| Scheme 2 Synthesis of chloroaldehyde 2a/2e. Reagents & conditions: (a) TBAHS, CH3I, CH2Cl2 for (1a) (b) (i) imidazole, DMF, TBDMSCl, 0 °C to r.t. (ii) NaH in DMF, BnBr (iii) TBAF in THF for (1b) (c) POCl3/DMF, 60 °C for 12 h. | ||
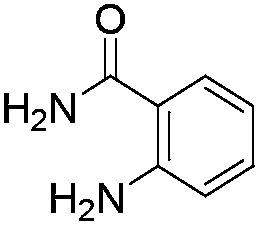
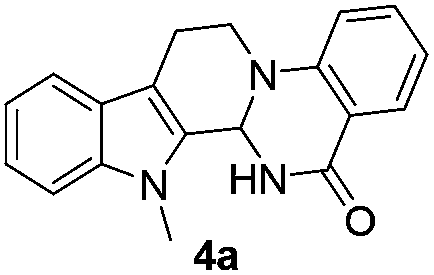
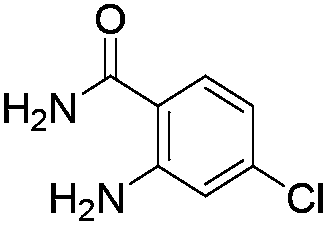
Initially, we attempted the coupling of 2a with anthranilamide (3) in DMF. The reaction was performed at different temperatures in various solvents and the results are presented in Table 1. The reaction was found to be sluggish at ambient temperature and the product 4a was isolated only in 35% yield after a long reaction time (12 h). By increasing the temperature from 25 to 120 °C, the desired product 4a was obtained in 90% yield (entry e, Table 1). Of various solvents like DMF, DMSO, and 1,2-dichlorobenzene tested, DMF gave the best results (Table 1).
| Entry | Solvent | Temp. °C | Time (h) | Yield (%) |
|---|---|---|---|---|
| a o-DCB = ortho-dichlorobenzene. | ||||
| a | DMF | 25 | 3 | 10 |
| b | DMF | 25 | 6 | 20 |
| c | DMF | 25 | 12 | 35 |
| d | DMF | 65 | 24 | 75 |
| e | DMF | 120 | 5 | 90 |
| f | DMSO | 25 | 12 | NR |
| g | DMSO | 120 | 12 | 55 |
| h | o-DCB | 120 | 12 | 50 |
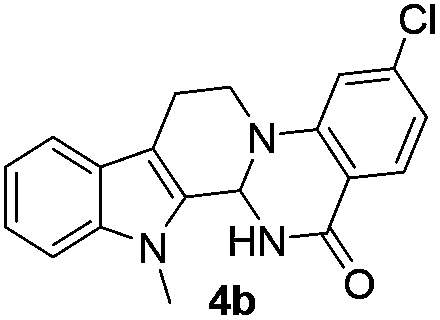
Under optimized conditions, the required product 4a was isolated in 90% yield. The structure of 4a was established by X-ray crystallography (Fig. 2).11
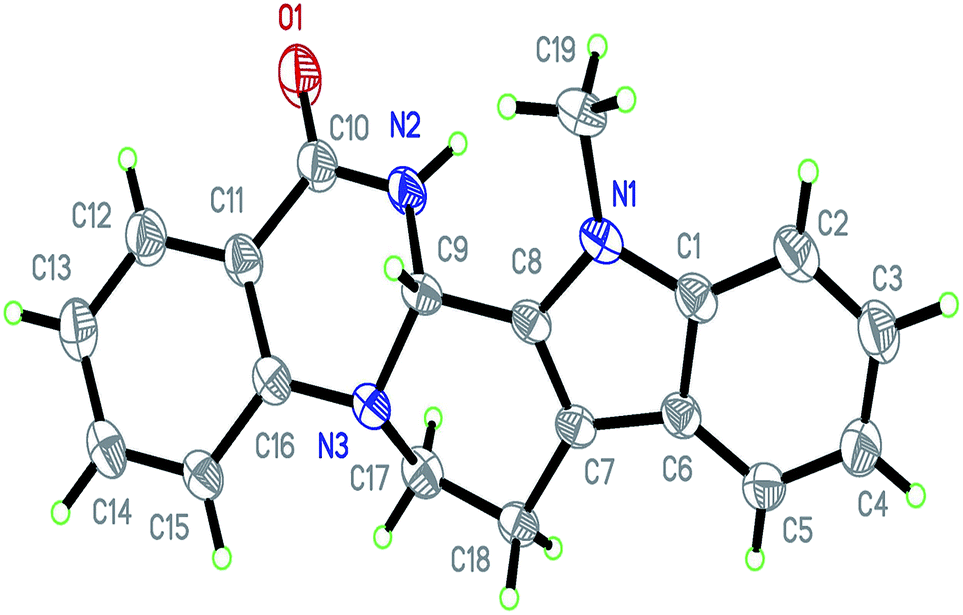 | ||
| Fig. 2 ORTEP diagram of 4a. | ||
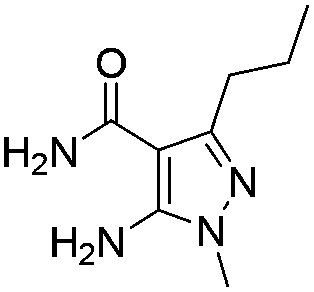
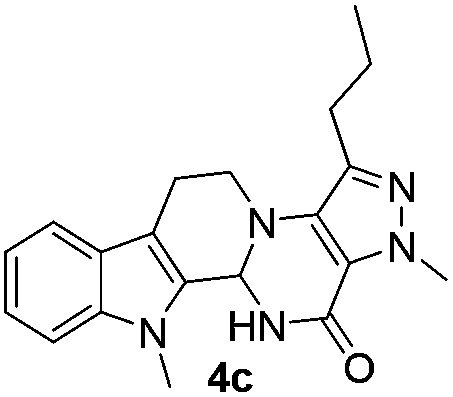
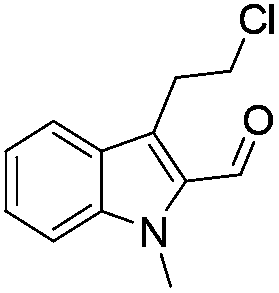
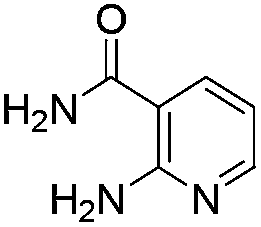
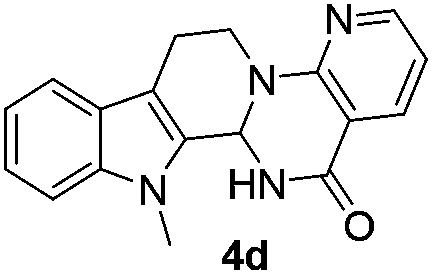
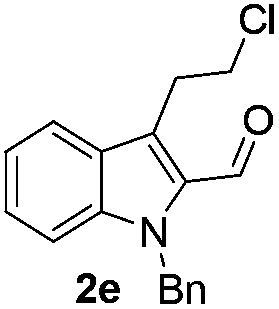
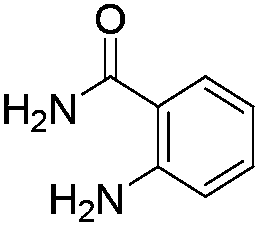
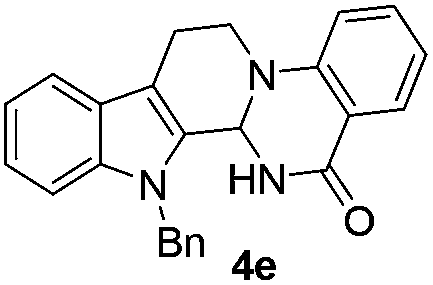
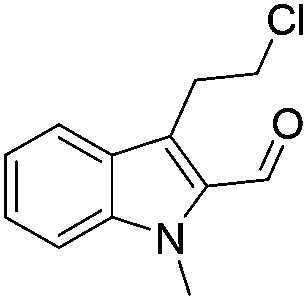
Similarly, the coupling of chloroaldehyde (2a) with 5-amino-1-methyl-3-propyl-1H-pyrazole-4-carboxamide afforded the product 4c in 87% yield (entry c, Table 2). Furthermore, the cyclization of 2a with 2-aminopyridine-3-carboxamide gave the respective product 4d in 85% yield (entry d, Table 2). Likewise, 1-benzyl-3-(2-chloroethyl)-1H-indole-2-carbaldehyde (2e) also participated well in this cyclization (entry e, Table 2).
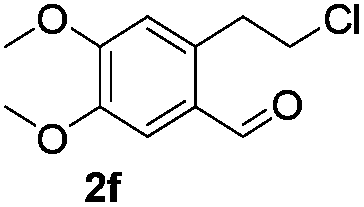
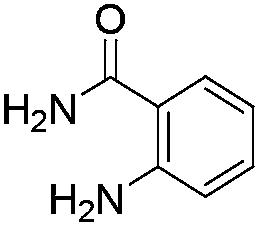
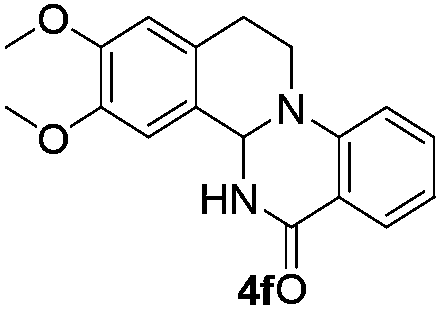
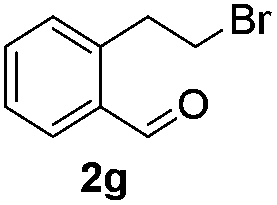
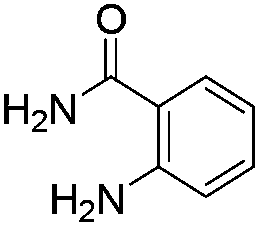
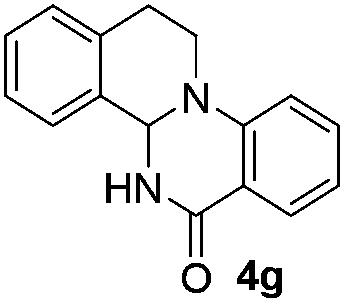
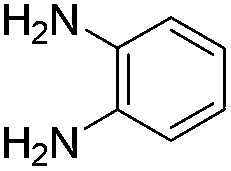
The scope of this method is further exemplified with 2-(2-chloroethyl)-4,5-dimethoxybenzaldehyde 2f12 with anthranilamide afforded the corresponding 4f in 89% yield (entry f, Table 2). In addition, 2-(2-bromoethyl)benzaldehyde (2g)13 also worked well for this conversion to produce the corresponding 4g in 86% yield (entry g, Table 2). Inspired by the above results, we extended this approach to the condensation of chloroaldehydes with o-phenylenediamines (5). Interestingly, this reaction proceeded well under similar conditions to furnish the corresponding benzimidazocarbolines (6) in good yields (Table 3).
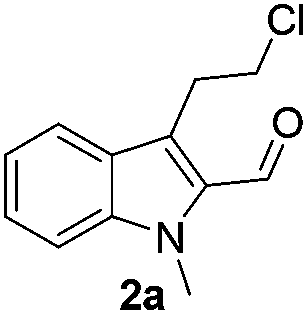
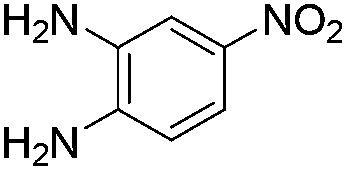
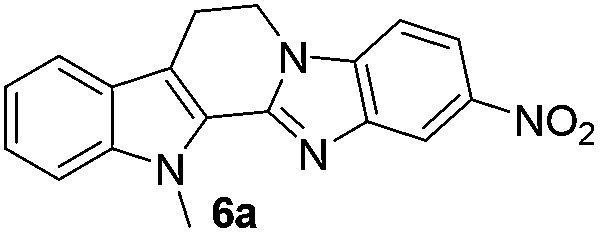
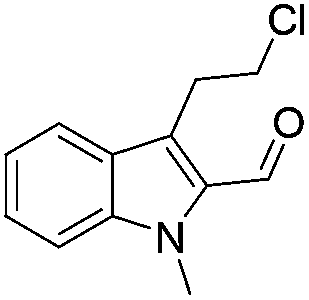
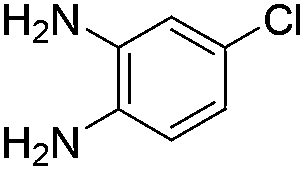
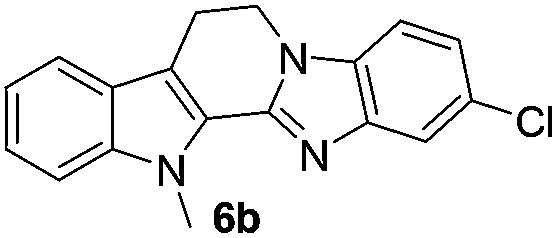
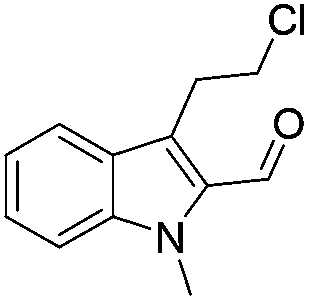
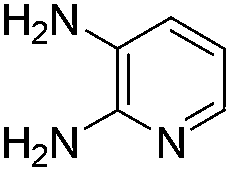
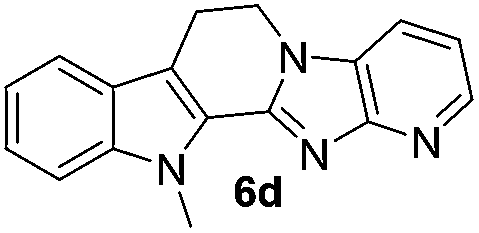
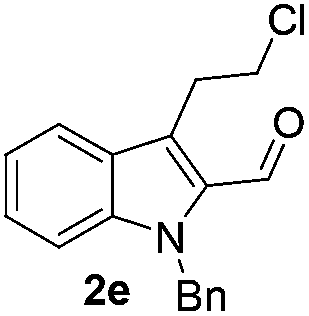
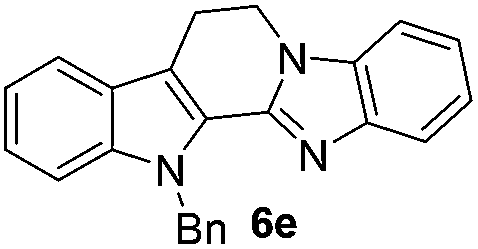
No significant effect of the substituents that are present on haloaldehyde (2), o-amino-carboxamide (3) or o-phenylenediamine (5) was observed under present reaction conditions. It is noteworthy to mentioned that only one regioisomer is formed, in which the substituent is meta to imine functionality as confirmed by 1H NMR spectrum (entries a and b, Table 3).14a This is due to electronic effects of the substituents. Furthermore, pyridine-2,3-diamine also afforded a single regioisomer as reported earlier in literature (entry d, Table 3).14b The reaction was quite successful with both N-methyl and N-benzyl chloroaldehydes. Thus this method is very useful for the synthesis of a diverse range of carbolines in a simple way. Mechanistically, the reaction is expected to proceed via the formation of iminium ion. This is attacked by the internal amido group to give the desired product (Scheme 3).
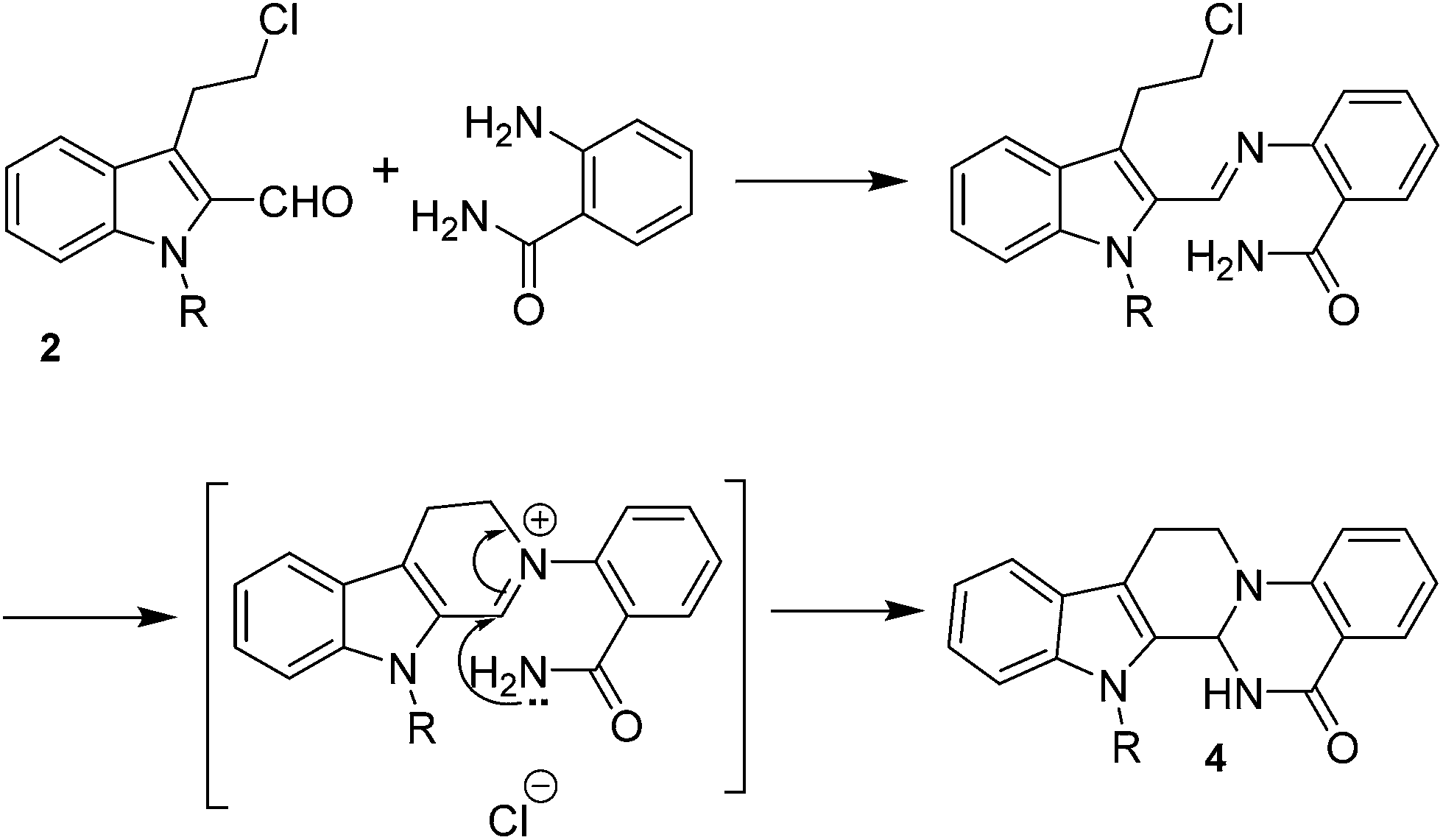 | ||
| Scheme 3 A plausible reaction pathway. | ||
In vitro anticancer activity
The end products were evaluated for their anti-proliferative activity against three human tumor cell lines; A549, HeLa and MCF-7. In order to examine the anti-proliferative effects, different concentrations of test compounds (1–100 μM) were incubated with three cell lines for 48 h along with the positive control (rutaecarpine) and examined the cell viability employing MTT assay.The results indicate that while rutaecarpine exhibited an IC50 value of 53.8 μM in HeLa cells, compound 4a exhibited 33.3 μM. However, for all other compounds in the series, the values obtained were more than 70 μM. In SK-N-SH cells, while the compounds 4e and 6e exhibited ∼92 μM, the values were nearly 50 μM for compounds 4d and 6d. The rest of the compounds in the series exhibited the IC50 values almost comparable to that of rutaecarpine (∼67 μM). However, in MCF-7 breast cancer cells, except for compound 6d (IC50 of 94 μM), all other compounds in the series including the positive control rutaecarpine exhibited the IC50 values much more than the maximum employed concentration i.e., 100 μM. The obtained values indicate that the compounds in the series are less cytotoxic, however, their precise physiological effects needs to be examined further (Table 4).
| Compound | HeLa | SK-N-SH | MCF-7 |
|---|---|---|---|
| 4a | 33.3 | 72.9 | 219 (>100) |
| 4b | 93 | 68.7 | 650 (>100) |
| 4c | 1323 (>100) | 70.9 | 474 (>100) |
| 4d | 89.1 | 53.8 | 1164 (>100) |
| 4e | 89.9 | 92.8 | 654 (>100) |
| 4f | 73.9 | 67.6 | 1831 (>100) |
| 4g | 297.6 (>100) | 70.6 | 283 (>100) |
| 6a | 247 (>100) | 72.9 | 198 (>100) |
| 6b | 319 (>100) | 69.3 | 378 (>100) |
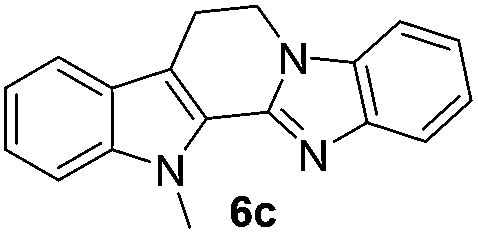
| 6c | 1012 (>100) | 70.9 | 754 (>100) |
| 6d | 294 (>100) | 54.1 | 94 |
| 6e | ND | 92.8 | 170 (>100) |
| Rutaecarpine | 53.8 | 67.2 | 749 (>100) |
In summary, we have developed a simple and convenient strategy for the synthesis of several analogues of rutaecarpine. This method also provides a direct access to benzimidazocarbolines and quinazolinoisoquinolines of biological importance.
Experimental
IR spectra were recorded on FT-IR spectrometer (KBr) and reported in reciprocal centimetres (cm−1). 1H NMR spectra were recorded at 600 MHz, 500 MHz, 300 MHz and 13C NMR at 150, 125 MHz, 75 MHz. For 1H NMR, tetramethylsilane (TMS) was used as internal standard (δ = 0) and the values are reported as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), and the coupling constants in Hz. For 13C NMR, CDCl3 (δ = 77.27) was used as internal standard and spectra were obtained with complete proton decoupling. Low-resolution MS and HRMS data were obtained using ESI ionization. Melting points were measured on micro melting point apparatus. Reaction progress was monitored by thin layer chromatography (TLC) on precoated silica gel GF254 plates and the spots were detected under UV light (254 nm).Materials and methods
Cell cultures, maintenance and evaluation of cytotoxicity
All the cell lines used in this study were purchased from the American Type Culture Collection (ATCC, United States). A549, HeLa and MCF-7 were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS in a humidified atmosphere of 5% CO2 at 37 °C. SK-N-SH neuroblastoma cells were cultured in Eagle's minimal essential medium (MEM) containing non-essential amino acids, 1 mM sodium pyruvate, 10 mg mL−1 bovine insulin, and 10% FBS. Cells were trypsinized when sub-confluent from T25 flasks/60 mm dishes and seeded in 96-well plates. The cytotoxicity of test compounds were evaluated based on their in vitro anti-proliferative effects in the above mentioned cell lines. The cell lines were grown in their respective media containing 10% FBS and were seeded into 96-well microtiter plates in 200 μL aliquots at plating densities depending on the doubling time of individual cell lines. The microtiter plates were incubated at 37 °C, 5% CO2, 95% air, and 100% relative humidity for 24 h prior to addition of experimental drugs. Aliquots of 2 μL of the test compounds (DMSO concentration was kept <1%) were added to the wells already containing 198 μL of media resulting in the required final concentrations of test compounds. For each compound, five concentrations (1, 10, 25, 50 and 100 μM) were evaluated and each was done in triplicate wells. Plates were incubated further for 48 h, and the experiment was terminated by the addition of 10 μL of 5% MTT and incubated further for 60 min at 37 °C. Later, the plates were washed and air-dried and bound stain was subsequently dissolved using 100 μL of DMSO. The absorbance was monitored in a multimode plate reader (PerkinElmer, Inspire model) at a wavelength of 560 nm. Percent viability was calculated on a plate by plate basis for test wells relative to control wells. The growth inhibitory effects of the compounds were analyzed by generating dose response curves as a plot of the percentage surviving cells versus compound concentration. The sensitivity of the cancer cells to the test compound was expressed in terms of IC50, the concentration of compound that produced 50% reduction in viability as compared to the control cells.General procedure for the preparation of (4)/(6)
To a solution haloaldehyde (2) (0.5 mmol) in anhydrous DMF (4 mL) was added o-amino-carboxamide (3) or o-phenylenediamine (5) (0.5 mmol) and the reaction mixture was stirred at 120 °C for 5–6 h. After completion, as indicated by TLC, the mixture was cooled to room temperature and poured drop wise while vigorous stirring into 30 mL of ice cold water. The precipitated solid was collected by filtration, washed with water, dried in vacuo to give the quinazolinocarboline or quinazolinoisoquinoline or benzimidazocarboline.Acknowledgements
B. V. S thanks CSIR, New Delhi for the financial support as a part of XII five year plan program under title ORIGIN (CSC-0108).Notes and references
- (a) Y. Asahima and K. Kashiwaki, J. Pharm. Soc. Jpn., 1915, 35, 1293 Search PubMed; (b) J. Bergman, in The Alkaloids, ed. A. R. Brossi, Academic, NewYork, 1983, vol. 21, p. 29 Search PubMed; (c) A. Ikuta, H. Urabe and T. Nakamura, J. Nat. Prod., 1998, 61, 1012 CrossRef CAS PubMed; (d) P. M. Joseph, Nat. Prod. Rep., 2001, 18, 543 RSC.
- (a) N. Shoji, A. Umeyama, T. Takemoto, A. Kajiwara and Y. Ohizumi, J. Pharm. Sci., 1986, 75, 612 CrossRef CAS; (b) Y. C. Kong, Adv. Pharm. Ther., 1982, 6, 239 CAS; (c) S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62, 9787 CrossRef CAS PubMed; (d) P. C. Sharma, G. Kaur, R. Pahwa, A. Sharma and H. Rajak, Curr. Med. Chem., 2011, 18, 4786 CrossRef CAS; (e) M. Demeunynck and I. Baussanne, Curr. Med. Chem., 2013, 20, 794 CAS.
- Y. C. Kong, S. Y. Hu, F. K. Lau, C. T. Che, H. W. Yeung, S. Cheung and C. C. Hwang, Am. J. Chin. Med., 1976, 4, 105 CrossRef CAS.
- (a) A. Kiss, J. Kokosi, R. Rotter and I. Hemecz, Tetrahedron, 2000, 56, 7987 CrossRef CAS; (b) N. Keumhan, M. S. Young, K. L. Sang, R. B. Sudeep, J. K. Mi, J. K. Yurngdong, W. K. Eunyoung and C. J. Tae, Arch. Pharmacal Res., 2011, 34, 119 CrossRef PubMed; (c) C.-S. Lee, C.-K. Liu, Y.-Y. Cheng and C.-M. Teng, Heterocycles, 2009, 78, 1047 CrossRef CAS; (d) X.-P. Qin, S.-Y. Zeng, D. Li, Q. Q. Chen, D. Luo, Z. Zhang, G. Y. Hu, H. W. Deng and Y. J. Li, J. Cardiovasc. Pharmacol., 2007, 50, 654 CrossRef CAS PubMed; (e) Y. W. Shin, E. A. Bae, X. F. Cai, J. J. Lee and D. H. Kim, Biol. Pharm. Bull., 2007, 30, 197 CrossRef CAS.
- S. Kim, S. H. Lee, E.-S. Lee, C.-S. Lee and Y. Jahng, Arch. Pharmacal Res., 2012, 35, 785 CrossRef CAS PubMed.
- (a) M. Adams, A. Mahringer, O. Kunert, G. Fricker, T. Efferth and R. Bauer, Planta Med., 2007, 73, 1554 CrossRef CAS PubMed; (b) J.-J. Chen, H.-Y. Fang, C.-Y. Duh and I.-S. Chen, Planta Med., 2005, 71, 470 CrossRef CAS PubMed.
- (a) L. M. Yang, S. J. Lin, L. C. Lin and Y. H. Kuo, Chin. Pharm. J., 1999, 51, 219 CAS; (b) L. M. Yang, C. F. Chen and H. L. Kuo, Bioorg. Med. Chem. Lett., 1995, 5, 465 CrossRef CAS; (c) G. R. Martins, H. B. Napolitano, L. T. F. M. Camargo and A. J. Camargo, J. Braz. Chem. Soc., 2012, 23, 2183 CrossRef CAS PubMed.
- S. P. Chavan and R. Sivappa, Tetrahedron Lett., 2004, 45, 997 CrossRef CAS PubMed.
- (a) G. Huang, D. Roos, P. Stadtmüller and M. Decker, Tetrahedron Lett., 2014, 55, 3607 CrossRef CAS PubMed; (b) S. H. Lee, J.-K. Son, B. S. Jeong, T.-C. Jeong, H. W. Chang, E.-S. Lee and Y. Jahng, Molecules, 2008, 13, 272 CrossRef CAS PubMed; (c) P. Frederic, C. Sebastien and D. Adam, Tetrahedron, 2011, 67, 5564 CrossRef PubMed.
- J. S. Yadav and B. V. S. Reddy, Tetrahedron Lett., 2002, 43, 1905 CrossRef CAS.
- ESI.†.
- (a) M. Sakamoto, D. Tanaka, H. Tsunoyama, T. Tsukuda, Y. Minagawa, Y. Majima and T. Teranishi, J. Am. Chem. Soc., 2012, 134, 816 CrossRef CAS PubMed.
- (a) M. Yamato, K. Washigaki, N. Qais and S. Ishikawa, Tetrahedron, 1990, 46, 5909 CrossRef CAS; (b) I. Coldham, S. Jana, L. Watson and N. G. Martin, Org. Biomol. Chem., 2009, 7, 1674 RSC.
- (a) M. A. Chari, D. Shobha and T. Sasaki, Tetrahedron Lett., 2011, 52, 5575 CrossRef PubMed; (b) R. P. Kale, M. U. Shaikh, G. R. Jadhav and C. H. Gill, Tetrahedron Lett., 2009, 50, 1780 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures and spectroscopic data for novel compounds, copies of 1H NMR, 13C NMR of novel compounds prepared are available. CCDC 1023835. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra14093a |
| This journal is © The Royal Society of Chemistry 2015 |