
Silver mediated duplex-type complexes of pyrimidinophanes and their acyclic counterparts†
Sergey N. Podyachev*a,
Alexey N. Masliyb,
Vyacheslav E. Semenova,
Victor V. Syakaeva,
Svetlana N. Sudakovaa,
Julia K. Voroninaa,
Vladimir T. Ivanova,
Andrey M. Kuznetsovb,
Edward L. Gogolashvilia,
Vladimir S. Reznika and
Alexander I. Konovalova
aA. E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center of Russian Academy of Sciences, Arbuzov str., 8, 420088, Kazan, Russia. E-mail: spodyachev@iopc.ru; Fax: +7 843 2731872
bKazan National Research Technological University, K. Marks str., 68, 420015, Kazan, Russia
First published on 28th January 2015
Abstract
Complexation of metal cations with acyclic and macrocyclic nucleobase derivatives containing uracil and 2-thiocytosine units linked by polymethylene spacers was studied. The investigated compounds have demonstrated high extraction selectivity for the Ag+ ion over a large number of competitive cations (Na+, Ca2+, Co2+, Ni2+, Cu2+, Zn2+, Cd2+). The structure and composition of Ag+ complexes have been investigated by a variety of physical-chemical methods. The crystal structures of three Ag+ complexes with 2-thiocytosine have been established by X-ray analysis. To refine the composition of the complexes formed in solution, the self-diffusion coefficients have been determined by NMR spectroscopy. For the estimation of the most reliable complex structures, the combined analysis of NMR data and DFT calculations was successfully applied. It was shown that the tendency of 2-thiocytosine moieties to form N1-nitrogen coordinated complexes and the presence of a polymethylene spacer between amine groups in the pyrimidinophanes provides the formation of Ag+ mediated duplex-type multi-component complexes.
Introduction
Some of the current investigations on DNA are often focused on the experimental and theoretical study of their interaction with metal ions.1 An interest in this problem is due to the fact that DNA metal complexes are promising objects for the creation of a new class of materials such as DNA-based wires and sensors capable of detecting various metal ions in an aqueous solution. The incorporation of transition metal complexes into DNA molecules could bring about materials in which the programmability and nanoscale rigidity of DNA are combined with the desired functionality of metal complexes.1–3 Besides, it is worth noting that the phenomenon of binding metal ions by pyrimidine base pairs in DNA duplexes also attracts attention and is the subject of contemporary investigations.4,5In this context, nucleobase complexes with metal ions can be considered as convenient systems for the analysis of the interaction of metal ions with nucleic acids. In addition, the attachment of nucleobases to the organic frameworks and the investigation of their complex formation properties could provide a useful strategy for the assembling of molecular building blocks and the design of artificial receptors.5,6 A lot of information on coordination of transition and noble metals with nucleobases,7–9 specifically with uracil,10 is available at present. However, the metal ion-binding properties of spatially-organized ligands possessing an acyclic or cyclic topology and containing several nucleic acid fragments have still not investigated and understood.
Recently, we have developed a series of acyclic and macrocyclic nucleobase derivatives with uracil and 2-thiocytosine moieties bridged with each other by polymethylene spacers.11,12 Some of the compounds investigated are represented in Scheme 1. The incorporation of 2-thiocytosine fragments into their structure resulted in the high affinity of the investigated compounds towards Ag+ ion. It should be noted also that in the synthesized compounds the N1-atom of the cytosine fragment is available for coordination, opposite to DNA, where it is covalently linked with the ribose. The presence of several donor centers in the synthesized nucleobase derivatives as well as the difference in their structural preorganization makes it possible to bring about various patterns of coordination for these compounds with metal cations, in particular, with the Ag+ ion. The establishment of the preferential coordination mode should be especially important for the elucidating the receptor properties of pyrimidinophanes and their acyclic counterparts.
 | ||
| Scheme 1 Structural formulae of the investigated compounds. | ||
Herein, we represent new results obtained for the binding properties of pyrimidine derivatives and the structures of their Ag+ complexes. Competitive extraction, X-ray crystallography, the combined analysis of NMR data and DFT calculations were used. Eventually, various models of the complex structures were discussed and compared.
Results and discussion
Competitive solvent extraction of metal cations
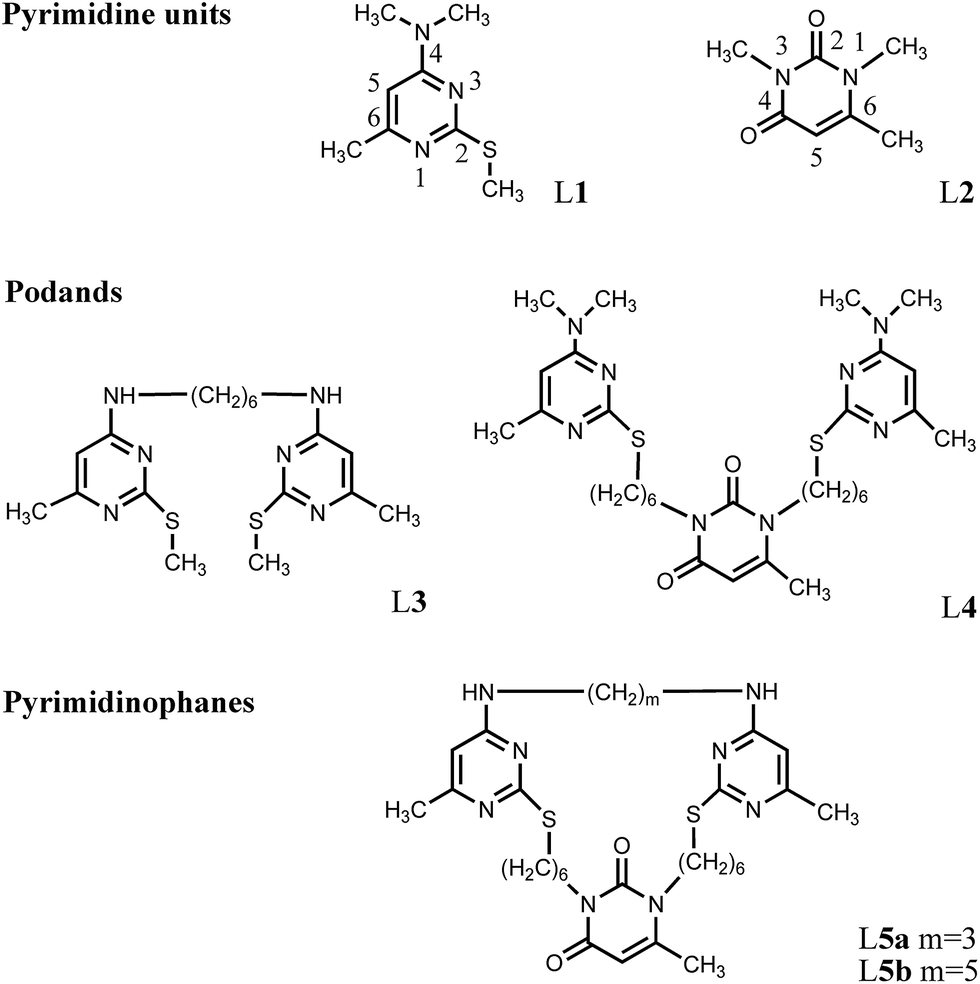
We have previously estimated the extraction ability of pyrimidine derivatives L1, L3, L4, L5a,b towards s- (Li+, Na+, K+, Cs+, Ca2+), d- (Co2+, Ni2+, Cu2+, Zn2+, Cd2+) and f- (La3+, Gd3+, Lu3+) metal ions by the noncompetitive extraction method which demonstrated a high affinity of the investigated compounds towards Ag+ ion.12 The extraction selectivity (S) for the Ag+ ion relative to the other studied metal ions in the case of macrocycle L5b reached the value S ≈ 50. These excellent results proved stimulating to proceed with our investigations. A competitive solvent extraction of the metal cations for L1, L3, L4 and L5a,b ligands has been carried out in the water–chloroform system. The concentrations of picric acid and metal cations (Na+, Ca+, Co2+, Ni+, Cu2+, Zn2+, Ag+ and Cd2+) in the aqueous phase were identical in all the experiments. The extraction percentages are summarized in Fig. 1.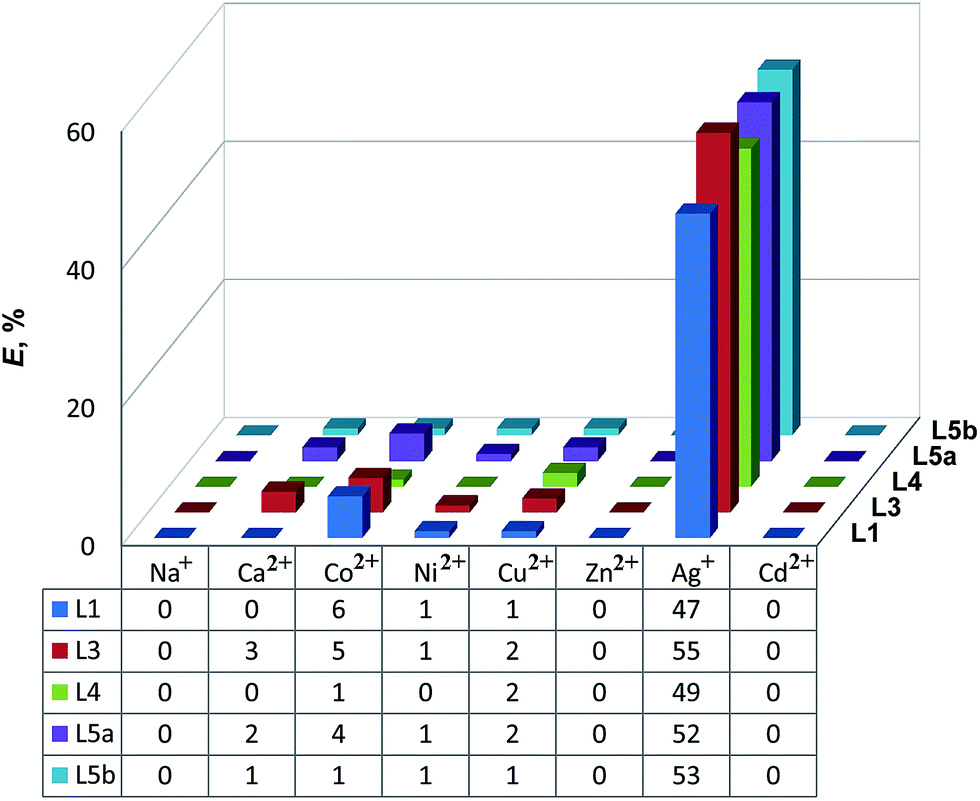 | ||
| Fig. 1 Extraction percentages (E%) of metal ions from water into CHCl3 at 298 K by L1, L3, L4, L5a and L5b. (CMz+ = 1 × 10−4 M; CL1 = 4 × 10−4 M; CL3,L4,L5a,L5b = 2 × 10−4 M; CHPic = 2 × 10−4 M). | ||
The results obtained have apparently demonstrated the good selectivity of the Ag+ ion recovery revealed by these ligands in the presence of a wide range of metal ions in the solution. For example, the extraction ratio Ag+/Men+ values determined for the compound L5b were up to ∼53, that agree with the data obtained for noncompetitive extraction selectivity. It has been clearly shown that the investigated compounds exhibit remarkable extracting properties towards silver ion. Furthermore, a significantly more effective complexation could be expected for the compounds possessing the podand and macrocyclic structures due to the cooperative effect of their binding sites. A more profound knowledge concerning the structures of the complexes and the donor centers involved in the coordination is necessary for a better investigation of the problem.
X-ray analysis of Ag+ complexes with L1
In order to clarify the predominant position of the Ag+ ion coordination by the thiopyrimidine derivatives, we have investigated the previously obtained complexes12 by X-ray diffraction analysis. Unfortunately, we only succeeded in obtaining of Ag·L12·Pic crystals, which were suitable for the X-ray study. Besides, in the present work the complexes of the L1 ligand with AgNO3 have been also obtained.The structure and molecular packing of the Ag·L12·Pic complexes in the crystal are shown in Fig. 2. It is clearly seen that the Ag+ ion forms a linear two-coordinate complex, which is approximately flat in the range of 0.0095(3) Å. In spite of the potential ability of the S atom to bind Ag+ cation, only the N1 atom of 2-thiocytosine is involved in the coordination with the Ag+ ion (Ag(1)–N(1) 2.122(2) Å and Ag(1)–N(13) 2.120(2) Å). In the crystals, all discrete units of the compound are linked through the mixed π⋯π stacking interactions: –(2-thiocytosine-2-thiocytosine-2-picrate)–. The distances between the centers of interacting rings are 3.69 Å and 3.79 Å. The dihedral angles formed by the planes are 4.28° and 4.79°, respectively. Under such packing, a short contact between the silver atom and the oxygen of the nitro group of picrate anion is realized (Ag(1)–O(40) 2.788(3) Å). The distance between these atoms is less than the sum of their van der Waals radii by 0.45 Å. It is worth noting that such type of π⋯π interactions is inherent for the double helix of a DNA and plays an important role in the stabilization of its structure.13
 | ||
| Fig. 2 Geometry (a) and molecular packing of Ag·2L1·Pic complex (b) in the crystal (hydrogen atoms are omitted for clarity). | ||
When using AgNO3 instead of the AgPic salt in a similar experimental condition, the crystals of Ag·L12·NO3 complex with analogous stoichiometry were obtained. As in the case of Ag·L12·Pic, the silver ion forms a two-coordinate linear complex (Ag(1)–N(1) 2.130(6) Å, Ag(1)–N(13) 2.129(6) Å), which is approximately flat in the range of 0.032(6) Å (Fig. 3a). 2-Thiocytosine molecules are also associated with each other through π⋯π stacking interactions: –(2-thiocytosine)– in the crystals of the complex. The distances between the centers of the interacting rings are approximately equal – (4.058(4) Å and 4.061(4) Å) and the dihedral angles between planes are 0.32° and 1.10°. However, it should be noted that the picrate anions in Ag·L12·Pic complexes form π⋯π stacks, but in the case of Ag·L12·NO3, the counter-ions are located in the channels between them (Fig. 3b).
 | ||
| Fig. 3 Geometry (a) and crystal packing (b) of Ag·L12·NO3. Counter anions are disordered over two positions with occupancies of 0.53 and 0.47. | ||
When the amount of AgNO3 was increased from 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 up to 1:1 of the mole ratio Ag:L1, the crystals of the complexes with another stoichiometry Ag2·L12·(NO3)2 were obtained. Similar to the complexes described above, the Ag(1) atom is coordinated with two 2-thiocytosine molecules via N1 nitrogen atom (Ag(1)–N(1) 2.140(3) Å, Ag(1)–N(13) 2.144(3) Å) (Fig. 4a). The second silver atom Ag(2) is also linked with the 2-thiocytosine fragments, but in this case by means of the S atoms of the ligands (Ag(2)–S(3) 2.626(1) Å, Ag(2)–S(4) 2.673(1) Å). Both of the NO3− anions are involved in the coordination with the Ag(2) cation. The attachment of the second silver atom is obviously provided by the stabilization of the complex structure due to that of NO3− anions playing the role of a bridge between the Ag+ ions (Ag(1)–Ag(2) = 3.0924(9) Å, Ag(1)–O(31) 2.645(5), Ag(2)–O(30) 2.293(4) Å). The second NO3− anion is bidentantly coordinated with Ag(2) (Ag(2)–O(27) 2.505(4), Ag(2)–O(26) 2.516(4) Å).
:1 up to 1:1 of the mole ratio Ag:L1, the crystals of the complexes with another stoichiometry Ag2·L12·(NO3)2 were obtained. Similar to the complexes described above, the Ag(1) atom is coordinated with two 2-thiocytosine molecules via N1 nitrogen atom (Ag(1)–N(1) 2.140(3) Å, Ag(1)–N(13) 2.144(3) Å) (Fig. 4a). The second silver atom Ag(2) is also linked with the 2-thiocytosine fragments, but in this case by means of the S atoms of the ligands (Ag(2)–S(3) 2.626(1) Å, Ag(2)–S(4) 2.673(1) Å). Both of the NO3− anions are involved in the coordination with the Ag(2) cation. The attachment of the second silver atom is obviously provided by the stabilization of the complex structure due to that of NO3− anions playing the role of a bridge between the Ag+ ions (Ag(1)–Ag(2) = 3.0924(9) Å, Ag(1)–O(31) 2.645(5), Ag(2)–O(30) 2.293(4) Å). The second NO3− anion is bidentantly coordinated with Ag(2) (Ag(2)–O(27) 2.505(4), Ag(2)–O(26) 2.516(4) Å).
 | ||
| Fig. 4 Geometry (a) and crystal packing (b) of Ag2·L12·(NO3)2 (hydrogen atoms are omitted for clarity). | ||
In these crystals, all the complexes are linked pairwise (Fig. 4b) due to the oxygen atoms of NO3− anions participating in the bifurcate binding of sulfur atoms of 2-thiocytosine molecules belonging to the neighboring complexes ((N–O(1)⋯S(3′) 3.180(3) Å and N–O(1)⋯S(4′) 3.196(3) Å). Besides, the oxygen atoms of the NO3− groups participate in additional CH⋯O interactions with the hydrogen atoms of the methyl groups of the adjacent complexes (the distances between the donor and acceptor are in the range of 3.382–3.476(7) Å). Owing to these CH⋯O interactions, the pairs of the complexes are linked in layers which are associated between themselves by the π⋯π interactions of 2-thiocytosine molecules. The distances between the centers of the interacting rings (4.176(3) Å, dihedral angle 7.1(2)°) and 4.146(3) Å, dihedral angle 15.3(2)°) exceed the similar spacing observed in the crystals of the Ag·L12·Pic complex by ∼0.4 Å.
Thus, the replacement of the Pic− chelating anion by that of NO3− can lead to the formation of a binuclear Ag2·L12·(NO3)2 complex with four- and hexa-coordinated Ag+ ions due to the intra-sphere coordination of the counter-ions and the participation of either of the two NO3− anions in the bridge binding of silver. Only in this complex, one of the silver ions is involved in the coordination with S atoms of the 2-thiocytosine molecules. In all other cases, the main coordination mode of the 2-thiocytosine ligands is provided by the binding of Ag+ ion via the N1 – nitrogen atom.
NMR Ag+ cation complexation studies
The estimation of ionophoric properties of pyrimidines, accomplished by the liquid extraction and NMR experiments, has demonstrated their high affinity towards Ag+ ions. Thus, it was established that the investigated pyrimidinophanes as well as their acyclic counterparts form stable 1:1 complexes with Ag+ ion both in polar media (CDCl3/DMSO-d6 = 1:1 (v/v) and CD3OD) and in a low-polarity solvent such as chloroform.12 The analysis of the Δδ1H and Δδ15N values obtained from NMR spectra and IR absorption bands of the ligands L1,3–5 and their complexes with AgPic testifies to the fact that 2-thiocytosine fragments entering these compounds are mainly responsible for the binding of metal ions. Moreover, NMR data indicate a possible involving of the same nitrogen heteroatoms of the 2-thiocytosine fragments in the coordination. Small changes of chemical shifts (Δδ = 0.07 ÷ 0.12 ppm) for the protons of the S–CH3 and S–CH2– groups in the NMR spectra of the complexes can be probably due to the non-participation of S atoms in the binding. The X-ray data obtained for the Ag·L12·Pic complex confirm this assumption as well. The coordination of Ag+ ion is realized only due to the N(1) atoms of the two 2-thiocytosine fragments.
Thus, it could be assumed that the L3–L5 compounds form complexes with AgPic following the same coordination mode. The anchoring of 2-thiocytosine fragments by the NH groups of polymethylene spacers in the podand L3 and pyrimidinophanes L5a,b is supposed to prevent a simultaneous involving of two 2-thiocytosine fragments in the metal binding and as a result lead to the bi-ligand coordination of the Ag+ ions. However, this assumption does not agree with the Ag:L (1:1) relationship experimentally established for the complexes.
It should be taken into account that the extraction data as well as the NMR titration data cannot distinguish between the formation of 1:1 or n:n complex stoichiometry, though this information is very important for the determination of the structures of these complexes. The investigated molecules have several coordination sites and typical NH proton-donor groups which are capable of generating associates even in polar solvents. To investigate the structures of the complexes formed in greater detail, we have performed the Fourier transform pulsed-gradient spin-echo (FT PGSE) NMR experiments. Currently, these methods are widely used for the investigation of a various kind of interactions, and make it possible, in particular, to learn how the cations and anions of transition metal salts interact in the solutions.14,15 In this connection, we have measured the self-diffusion coefficients (Ds) both for the solutions of free ligands (L1, L3, L4 and L5a,b) at various concentrations as well as for their complexes with Ag+Pic− (Table 1). The complexes with higher molecular weights appeared to possess the lower Ds-values.
| Compound | CL, mM | CAgPic, mM | Ds, 10−10 m2 s−1 | V(L + Ag+Pic−)/V(Lfree) |
|---|---|---|---|---|
| a Particle volume changes at the increased concentration of free ligands. | ||||
| L1 | 1 | 6.13 | ||
| L1 | 30 | 6.17 | 1.0a | |
| L1 + Ag+ | 30 | 15 | 4.64 | 2.3 |
| L3 | 1 | 3.57 | ||
| L3 | 30 | 3.14 | 1.5a | |
| L3 + Ag+ | 30 | 30 | 1.42 | 16.0 |
| L4 | 1 | 3.28 | ||
| L4 | 30 | 3.36 | 1.0a | |
| L4 + Ag+ | 30 | 30 | 3.06 | 1.2 |
| L5a | 1 | 2.88 | ||
| L5a | 10 | 2.69 | 1.2a | |
| L5a + Ag+ | 10 | 10 | 2.26 | 2.1 |
| L5b | 1 | 2.40 | ||
| L5b | 10 | 2.38 | 1.0a | |
| L5b + Ag+ | 10 | 10 | 1.78 | 2.4 |
An increase of the L1 and L4 ligand concentrations in the solution does not actually lead to a change in the sizes of the particles. However, in the case of compound L3 we observed the volume of particles to be about 1.5 times greater, which indicates a dimerization of molecules in the solution. The presence of the NH groups in L3 molecules is, obviously, favorable for their aggregation. An addition of Ag+Pic− to the solution of ligand L1 resulted in an increase in the volume of particles up to 2.3 times, which is in agreement with the formation of biligand Ag+ complexes in the investigated solutions. It should be noted that the structure of the complex isolated from the methanol solution and established by X-ray analysis had the same composition and was considered above. An addition of one equivalent of Ag+Pic+ to the solution of compound L3 leads to a sharp enhancement (∼16 times) in the volume of particles. The formation of such large particles is obviously due to the polymer structure of the complexes in solution.
In the case of podand L4, the increase in the volume of particles was not practically observed, which testifies to the formation of mono-ligand complexes with Ag+ ions for this compound. This fact is in accordance with the stoichiometry of the complexes 1:1 established by the extraction method, NMR titration data as well as the elemental analysis of the complexes in a solid.12 It is interesting to note that our attempts to obtain complexes with AgNO3 in MeOH solutions resulted in a fast formation (∼5 minutes) of a gel substance which remained stable for several months. After lowering the concentration of the ligand from 1.5 wt% to 0.75 wt%, the gel formation process was completed after a day. Thus, by varying a solvent and/or a counter ion, it proved to be possible to control the supramolecular structure of Ag+ complexes in a solution. A search for new gelators and the conditions of their formation is an extremely promising field of research16,17 and requires a self-sufficient investigation.
In the case of complex formation with the pyrimidinophanes L5a and L5b, a double increase in the volume of particles was observed. However, it was previously established that compounds L5a and L5b bind Ag+ ions in the 1:1 relationship between the metal and ligand.12 Both of these facts are obviously due to the dimer structure of the Ag+ complexes (2Ag+:2L5a,b) for these ligands.
Thus, it can be concluded that an addition of Ag+ to the solutions of pyrimidinophanes L5a and L5b and their acyclic counterpart L3 mediates the formation of duplex-type complexes. The presence of an alkyl bridge between the amine groups of 2-thiocytosine fragments prevents their simultaneous intra-molecular coordination with the Ag+ ion and is a necessary condition for the formation of such type of complexes.
For the purpose of obtaining further information about the structures of complexes, we have performed quantum-chemical calculations using the density functional theory (DFT) methods. The data obtained have been compared with the data established by the NMR spectroscopy.
Theoretical studies
The process of extracting metal cations from aqueous to organic phase is accompanied by z picrate anions and n neutral organic ligands (L) and can be described by eqn (1):| M(aq)z+ + zPic(aq)− + nLorg⇆[Mz+LnPicz−](org) | (1) |
In this case, Mz+, Pic−, L, [Mz+LnPicz−] denote the metal ion, picrate anion, ligand and ion-pair metal complex, respectively. The subscripts aq and org mean that the species exist in aqueous or organic phase.
Chloroform has been used as an organic phase. Probably, picrate anion does not coordinate the Ag+ ion during complex formation. This fact is in agreement with the X-ray analysis data obtained for silver picrate complexes of the ligand L1. It should be noted that the extraction process is considerably affected by the solvation of species. In this connection, we used a linear aqua complex [Ag(H2O)2]+ for the determination of thermochemical characteristics of the reactions proceeding with the Ag+ ions in water solutions. According to the quantum-chemical calculations, the formation of such a complex is most probable.
Previously obtained extraction data have evidenced that the interaction of Ag+ with L1 leads to the formation of bi-ligand complexes.12 A complex of similar stoichiometry has been isolated from the methanol solution after the interaction of AgPic with two mole equivalents of this ligand. Taking these comments into account, the formation of the [Ag(L1)2]+ complex can be described by the following equation:
| [Ag(H2O)2](aq)+ + 2L1(org) = [AgL12](org)+ + 2H2O(aq) | (2) |
The structures of ligand L1 related to the cis- and trans-conformations are specified only due to the location of SCH3 groups in the plane of pyrimidine ring and slightly differ from each other in their energy (ΔHo298 < 0.01 kcal mol−1) (see Fig. 5a and b). Therefore, the existence of these conformers in solution is equally probable. Each of these conformers can coordinate a silver cation by one of the nitrogen atoms (N1 or N3) as well as by a sulfur of 2-thiocytosine molecule. The optimization of the suggested complex structures has yielded two isomers. In one of the structures, the coordination is realized through the nitrogen N3, in another one – via the N1 atoms (Fig. 5c and d and Table 1). Sulfur atoms are not obviously involved in the binding process (R(Ag–S) > 3Å). The formation of the N1-coordinated complex is accompanied by the largest gain in energy (ΔHo298([AgL1(N3)2]+) = −21.82 kcal mol−1 and ΔHo298([AgL1(N1)2]+) = −9.64 kcal mol−1).
 | ||
| Fig. 5 The optimized structures of (a) cis- and (b) trans-conformers of L1 and [AgL12]+ complex with (c) N3–Ag+–N3′ and (d) N1–Ag+–N1′ coordination modes. | ||
Table 2 represents experimental and calculated values of the chemical shift differences Δδ1H (15N) both for free ligands and for their Ag+ complexes. The average deviations of these values (Δavδ1H(15N)) are given in Table 2 as well
| Δavδ1H(15N) = Σ[Δδ1H(15N)exp − Δδ1H(15N)calc]/n | (3) |
| Composition (coordination mode) | Figure | ΔHo298 | Epreorg | Δδ1H | Δavδ1H | Δδ15N | Δavδ15N | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S–CHn | C5–H | C6–CH3 | NH | N4(CHn)m | N1 | N3 | N4 | ||||||
| a Equimolar mixtures of Ag+ ion and ligand L in CDCl3–DMSO-d6 = 1:1 (v/v) at 303 K.b No data.c Per one Ag+ ion. |
|||||||||||||
| Ag+ + 2L1a | 0.11 | 0.23 | 0.18 | 0.08 | −31 | —b | —b | ||||||
| [AgL1(N3)2]+ | 5c | −9.64 | 3.03 | 0.60 | 0.50 | 0.20 | −0.13 | 0.25 | 1.1 | −31.7 | 0.1 | 32.1 | |
| [AgL1(N1)2]+ | 5d | −21.82 | 2.51 | 0.15 | 0.20 | 0.15 | 0.07 | 0.03 | −47.6 | 1.7 | 15.5 | 16.6 | |
| Ag+ + L3a | 0.07 | 0.16 | 0.13 | 0.83 | 0.11 | −33 | —b | 10 | |||||
| [AgL3(N3,N3)]+ | 6b | −2.64 | 9.79 | 0.30 | 0.50 | 0.30 | −0.20 | −0.05 | 0.39 | 6.0 | −38.5 | 4.1 | 22.5 |
| [AgL3(N3,N3)]+ | 6c | −1.74 | 9.70 | 0.4 | 0.5 | 0.4 | 1.3 | −0.15 | 0.33 | 6.2 | −36.5 | 8.1 | 20.6 |
| [AgL3(N3,S)]+ | 6d | −2.74 | 7.00 | 0.4 | 0.2 | 0.1 | 0 | −0.05 | 0.28 | 3.4 | −43.5 | 3.6 | 21.4 |
| [Ag2L3(N1,N1)2]2+ | 6e | −19.74, (−9.87)c | 7.58 | 0.1 | 0.4 | 0.1 | 0.9 | 0.05 | 0.09 | −47.9 | 3.7 | 11 | 8.0 |
| [Ag2L3(N1,N1)2]2+ | 6f | −18.92, (−9.46)c | 6.43 | 0.1 | 0.3 | 0.1 | 1.00 | 0.05 | 0.09 | −46.7 | 6.1 | 11.5 | 7.6 |
| [AgL3(N1)2]+ | 6g | −17.24 | 2.47 | 0.2 | 0.3 | 0.1 | 0.8 | 0.15 | 0.07 | −47.9 | 2.1 | 11.5 | 8.2 |
| [Ag2L3(N1,N1)L3(N1)2]2+ | 6h | −28.76, (−14.38)c | 3.31 | 0.2 | 0.3 | 0.15 | 0.8 | 0.15 | 0.07 | −47.7 | 0.9 | 11.8 | 8.3 |
| Ag+ + L4a | 0.12 | 0.29 | 0.2 | 0.11 | −33 | −18 | —b | ||||||
| [AgL4(N1,N3)]+ | 7b | −10.77 | 7.08 | 0.30 | 0.30 | 0.55 | 0.20 | 0.16 | −43.5 | −42.7 | 16.4 | 17.6 | |
| [AgL4(N1,N3)]+ | 7c | −11.15 | 7.45 | 0.30 | 0.30 | 0.25 | 0.19 | 0.08 | −42.7 | −42.5 | 16.1 | 17.1 | |
| [AgL4(N3,N3)]+ | 7d | −4.84 | 8.09 | 0.43 | 0.40 | 0.12 | 0.23 | 0.15 | 3.2 | −42.0 | 2.8 | 30.1 | |
| [AgL4(N1,N1)]+ | 7e | −17.52 | 7.07 | 0.30 | 0.30 | 0.22 | 0.23 | 0.08 | −43.1 | 2.0 | 14.0 | 15.1 | |
| Ag+ + L5aa | 0.09 | 0.14 | 0.12 | 0.6 | 0.14 | —b | —b | —b | |||||
| [AgL5a(N3,N3)]+ | 8b | 3.79 | 14.94 | 0.13 | 0.40 | 0.20 | −0.80 | 0.2 | 0.37 | 6.6 | 33.4 | −4.4 | |
| [AgL5a(S,S)]+ | 8c | 6.68 | 15.24 | 0.13 | 0.10 | −0.10 | −0.60 | −0.2 | 0.37 | −5.3 | 32.2 | −10.3 | |
| [AgL5a(S,N3,S)]+ | 8d | −4.57 | 10.10 | 0.63 | 0.40 | 0.20 | −0.20 | −0.35 | 0.43 | 7.1 | 33.6 | −3.8 | |
| [Ag2L5a(N1,N1)2]2 | 8e | −10.08, (−5.04)c | 14.46 | 0.13 | 0.20 | 0.20 | −0.25 | 0 | 0.23 | −35.9 | 7.0 | 14.0 | |
| [Ag2L5a(N1,N1)2]2+ | 8f | −8.34, (−4.17)c | 17.22 | −0.08 | 0.10 | 0.10 | 0.05 | 0.05 | 0.17 | −33.1 | 7.5 | 12.8 | |
It is clearly seen that the minimal deviation between the calculated and experimental chemical shift values is observed for N1, N1′-coordinated complexes represented in Fig. 5d.
The conclusions concerning the N1-coordination of Ag+ ion by L1, that were drawn on the basis of quantum-chemical calculations of the thermal effect of the complex formation process as well as the analysis of the experimental and calculated NMR chemical shifts of the compounds, are in good agreement with the X-ray data for the complex [AgL12]+Pic− (Fig. 2). The main difference between the structures of the investigated complexes consists in the mutual orientation of the 2-thiocytosine rings. The turn of the rings in the complex structure was revealed with the use of quantum-chemical calculations and is obviously provided by the repulsion of the CH3–S– and CH3– groups belonging to the adjacent 2-thiocytosine molecules. In a crystal, the packing effect leads to the flattening of the complexes, which is clearly shown by the X-ray data. Thereby, an approach based on the combined analysis of NMR data and DFT calculations can be successfully applied for the estimation of the most reliable complex structures.
The incorporation of two 2-thiocytosine fragments into the structure of a podand (compounds L3 and L4) results in an increase in the non-competitive extraction efficiency for Ag+ ion. The enhancement of the extraction process is obviously caused by the participation of both 2-thiocytosine moieties in the binding of a metal ion. The 1H NMR-titration performed earlier for the compound L4 has revealed that the uracil fragment is not involved in the coordination. According to the extraction and NMR data, the podands L3 and L4 as well as the pyrimidinophanes L5a and L5b form the complexes of a similar stoichiometry 1:1 (Ag+:L).
The consideration of the L4 ligand structure indicates the possible binding of Ag+ ion by N1 or N3 atoms of 2-thiocytosine fragments. Besides, sulfur atoms can also participate in the metal coordination. In the case of compound L3, the hexamethylene spacer prevents a simultaneous coordination of Ag+ ion by both N1 nitrogen atoms of the 2-thiocytosine fragments of the molecule. The simultaneous coordination of silver ions in the complex [AgL3]+ with the participation of two fragments of the ligand can be brought about only by means of their N3 and/or S-donor atoms. However, an addition of AgPic to the solutions of L3 and L4 results in a similar change of the NMR chemical shifts of the 2-thiocytosine fragments (Table 2). This fact can testify to a similar metal-coordination mode revealed by these ligands. On the other hand, according to the NMR data, the particle volume changes determined for [AgL3]+ in the CDCl3/DMSO-d6 solutions indicate the polymer structure of this complex (Table 1) in contrast to the Ag+ complexes formed by the L1 and L4 ligands.
In this connection, the realization of both the mononuclear, and polymer type of complexes of the ligand L3 (Fig. 6a) was considered. The formation of mononuclear complexes is described by the following equation:
| [Ag(H2O)2](aq)+ + L3(org) = [AgL3](org)+ + 2H2O(aq) | (4) |
 | ||
| Fig. 6 The optimized structures of (a) compound L3 and its complexes: [AgL3]+ with the (b) N3–Ag+–N3 (cis/cis oriented NH groups), (c) N3–Ag+–N3 (trans/cis oriented NH groups), (d) S–Ag+–N3 (trans/trans oriented NH groups) coordination modes; [Ag2L32]2+ with the (e) N1–Ag+–N1 (cis/cis oriented SCH3 groups) and (f) trans/trans oriented SCH3 groups; (g) [AgL32]+ and (h) [Ag2L33]2+. Only hydrogen atoms of NH groups are shown. | ||
For mononuclear complexes, as mentioned above, the formation of chelate structures with the coordination of metal ions via N3 and/or S atoms of the ligand appears to be possible (Fig. 6b–d). The cis- and trans-orientation of amine hydrogen atoms in the pyrimidine fragments can also affect the coordination process. The optimization of the complex model structures has resulted in the N3, N3′-coordinated structures which differ only by cis/cis (Fig. 6b) and cis/trans (Fig. 6c) orientation of the H4–N4–C4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N3 fragments. In the case of trans/trans orientation of the NH groups, the structure with N3, S-coordination of the Ag+ ion has been obtained (Fig. 6d).
N3 fragments. In the case of trans/trans orientation of the NH groups, the structure with N3, S-coordination of the Ag+ ion has been obtained (Fig. 6d).
Obviously, the coordination with the involvement of N1 atoms of 2-thiocytosine fragments is possible only in the case of a dimer complex. Its formation would occur in accordance with the following reaction:
| 2[Ag(H2O)2](aq)+ + 2L3(org) = [Ag2L32](org)2+ + 4H2O(aq) | (5) |
Optimized [Ag2L32]2+ complex structures (Fig. 6e and f) differ by their mutual location of the S–CH3 groups of ligand relative to the Ag+ ions and possess close ΔHo298 values.
In the case of the polymer complex [AgnL3n+1] described by a formal stoichiometry 1:1 at the large n, the Ag+ ion coordination can be realized through the N1 atom of the 2-thiocytosine fragment, similarly to that of the dimer complex. To simulate the structure of the polymer complex and estimate the thermal effect of its formation, the quantum-chemical calculations of its structural units [AgL32]+ and [Ag2L33]2+ have been performed (Fig. 6g and h). The formation of these complexes can be described by the following reaction:
| n[Ag(H2O)2](aq)+ + (n + 1)L3(org) = [AgnL3(n+1)](org)n+ + 2nH2O(aq) | (6) |
From the comparison of the calculated ΔHo298 values (Table 2), it was concluded that the formation of mononuclear N3, N3′-coordinated complexes [AgL3]+ would appear to be a rather unfavorable process (ΔHo298 = −1.74 and −2.64 kcal mol−1). A noticeable energy gain is observed in the case of the formation of N1, N1′-dimer complex [Ag2L32]2+ (ΔHo298 = −9.87 and −9.46 kcal mol−1) and especially for N1, N1′-coordinated complexes having a chain structure [AgL32]+ (−17.24 kcal mol−1), [Ag2L33]2+ (−14.38 kcal mol−1). Therefore, the most energy profitable process is provided by the formation of complexes with a chain structure. It is worth noting that similar energy changes are observed when the ligand preorganization energy in these complexes is taken into account (see Epreorg in Table 2). Their formation proceeds with a minimal energy consumption, due to the ligand preorganization process.
The deviations of the calculated and experimental values Δδ1H(15N) in the spectra of the silver complex and free ligand L3 (Table 2) become essentially lower from N3,N3′-mononuclear (Δavδ1H = 0.33–0.39 ppm, Δavδ15N = 20.55–22.45 ppm) to the N1,N1′-coordinated polynuclear (Δavδ1H = 0.08–0.09 ppm, Δavδ15N = 7.95–8.25 ppm) complexes. This well correlates with conclusions drawn from thermochemical calculations.
As mentioned above, the coordination of the podand L4 with Ag+ ions does not lead to the formation of polynuclear complexes, in contrast to the case with the ligand L3. Therefore, we have considered the possibility of the formation of a mononuclear [AgL4]+ complex with the coordination of Ag+ ion both via N1 and the N3 nitrogen atoms of the 2-thiocytosine moieties. According to the quantum-chemical calculations, L4 possesses an expanded structure (Fig. 7a). The presence of the uracil fragment in the bridge of molecule leads to the asymmetry of the podand structure and provides an existence of two complex isomers with the N1,N3-ligand coordination mode (complex [AgL4(N1,N3)]+). In this connection, the formation of four possible isomers of chelate complexes [AgL4]+ has been considered.
 | ||
| Fig. 7 The optimized structures of (a) compound L4 and its complexes: [AgL4]+ with the (b) N1–Ag+–N3, (c) N3–Ag+–N1, (d) N3–Ag+–N3 and (e) N1–Ag+–N1 coordination modes. | ||
The formation of these Ag+ complexes is described by the following reaction:
| [Ag(H2O)2](aq)+ + L4(org) = [AgL4](org)+ + 2H2O(aq) | (7) |
All of the four chelate complex isomers appeared to be stable (Fig. 7b–e).
In terms of energy and in accordance with thermochemical data, the formation of the complex with the N1,N1-coordination of silver ion looks to be more profitable (Fig. 7e). Coordination with the participation of N3 nitrogen atoms of the ligand L4 as well as the formation of N3,N3′-coordinated complex [AgL12]+ seems to be a substantially less favorable processes (ΔHo298([AgL1(N1,N1′)2]+) − ΔHo298([AgL1(N3,N3′)2]+) = 12.18 kcal mol−1 and ΔHo298([AgL4(N1,N1)2]+) − ΔHo298([AgL4(N3,N3)]+) = 12.68 kcal mol−1). When the considered complexes are formed, the values of the preorganization energy for the isomers of podand L4 are approximately equal, and therefore, in contrast to the case with the ligand L3, they do not significantly affect the preference in the formation of complexes [AgL4]+ (see Epreorg in Table 2).
The complex [AgL4(N3,N3)]+ (Fig. 7d) with the lowest stability reveals a maximum of deviations in Δavδ15N (30.1 ppm) and Δav1H (0.15 ppm) values (Table 2). Whereas, the most thermo-chemically stable [AgL4(N1,N1)]+ complex (Fig. 7e) gives the lowest deviations of the experimental and calculated NMR parameters (Δavδ1H = 0.08 ppm, Δavδ15N = 15.05 ppm). All these results confirm the conclusions drawn from thermo-chemical calculations.
The fact that both 2-thiocytosine fragments are connected by two polymethylene bridges is specific for pyrimidinophane structure. One of the bridges of the molecule includes a methyl uracil fragment. The structural optimization of macrocycle L5a has resulted in an expanded structure (Fig. 8a). Based on the extraction and 1H NMR titration data, it was previously established that pyrimidinophane L5a forms Ag+ complexes of the 1:1 stoichiometry.12 Obviously, the formation of mononuclear complexes [AgL5a]+ with the participation of both 2-thiocytosine fragments of the ligand can be realized only when the N3 nitrogen and/or S sulfur atoms of these fragments are involved in the coordination process. The self-diffusion coefficients obtained from the NMR experiments (Table 1) made it possible for us to assume the formation of dimer complexes [Ag2L5a2]2+ as well. The formation of mononuclear and dimer complexes can be described by the following reactions:
| [Ag(H2O)2](aq)+ + L5a(org) = [AgL5a](org)+ + 2H2O(aq) | (8) |
| 2[Ag(H2O)2](aq)+ + 2L5a(org) = [Ag2L5a2](org)2+ + 4H2O(aq) | (9) |
 | ||
| Fig. 8 The optimized structures of (a) compound L5a and its complexes: [AgL5a]+ with the (b) N3–Ag+–N3, (c) S–Ag+–S, (d) N3,S–Ag+–S coordination modes; isomers of complex [Ag2L5a2]2+ with the (e) N1,N1–Ag+–N1′,N1′ (trans) and (f) N1,N1–Ag+–N1′,N1′(cis) coordination modes. Only hydrogen atoms of NH groups are shown. | ||
The thermochemical data (Table 2) have clearly shown that the structures obtained for mononuclear complexes with N3,N3- and S,S-bidentant coordination (Fig. 8b and c) have a positive enthalpy of formation and, therefore, such a type of complexes in the solution is unlikely. Only the S,S,N3-tridentant complex (Fig. 8d) is stable. Besides, the formation of the N1,N1,N1′,N1′-coordinated binuclear complexes with cis- or trans-ligand orientation in the structure of the [Ag2L5a2]2+ complex (Fig. 8e and f) is accompanied by the largest gain in ΔHo298 among all the calculated structures.
The lowest deviations of the Δavδ1H (Table 2) values were observed for both dimer complexes. Based on a set of experimental and calculated data, it can be concluded that the complexes with the N1-coordination of the 2-thiocytosine fragments are formed in these cases as well. The realization of such a coordination mode becomes possible only in the case of the formation of the duplex-type complexes by pyrimidinophanes.
In general, for all the investigated pyrimidinophanes as well as for their acyclic counterparts, the coordination of silver ion is brought about by means of the N1 nitrogen atoms of 2-thiocytosine fragments, although, hypothetically, it can occur in a different way. The presence of a polymethylene spacer between the amine groups, preventing the N1,N1-coordination of Ag+ ion by the molecule of podand L3 or pyrimidinophane L5, leads to the formation of dimer or polymer complexes by these ligands. In the case of podand L3, the formation of Ag+ complexes with a chain structure has been observed, but for pyrimidinophane L5 the dimer structure of Ag+ complexes is preferable. Therefore, it is interesting to clarify the possible reason for such N1 coordination selectivity.
The charge distributions obtained by the quantum chemical calculations for the nitrogen and sulfur atoms of 2-thiocytosine fragments are represented in Table 3. All free ligands have a rather high positive charge on their sulfur atoms (0.39–0.47 a.u.), which results in the repulsion of the metal cations. The coordination of silver ions is possible only with the negative electron-donor nitrogen atoms (N1, N3 and N4). However, the N4-coordination is also hardly probable because of the steric hindrance provided by the alkyl substitutes. In addition, the negative charge on the N4 atom is almost twice lower in comparison with the N1 and N3 charges for free ligands. Each one of the heteroatoms, N1 or N3, can form a coordination bond with silver ion. In most cases, the charge at the N1 atom is insignificantly more negative in comparison with the N3 charge (∼0.01 a.u.). This fact can only indirectly indicate the preferable involvement of the N1 atom in the coordination process as compared to N3.
| Compound | Figure | N1 | N3 | N4 | S |
|---|---|---|---|---|---|
| a Charges are given only for atoms of 2-thiocytosine fragments participating in the coordination with Ag+ ion. Only one value of charge is represented when the charges on the same atoms in different fragments are very close (≤0.01 a.u.).b The former shows the charge on the atoms of 2-thiocytosine fragment locating on the side of carbonyl group (C4O) of the uracil bridge. |
|||||
| L1(cis) | 5a | −0.203 | −0.213 | −0.122 | 0.390 |
| L1(trans) | 5b | −0.218 | −0.203 | −0.121 | 0.395 |
| [AgL1(N3)2]+ | 5c | −0.176 | −0.201 | −0.101 | 0.485 |
| [AgL1(N1)2]+ | 5d | −0.207 | −0.182 | −0.94 | 0.459 |
| L3 | 6a | −0.218 | −0.200 | −0.080 | 0.397 |
| [AgL3(N3,N3)]+ | 6b | −0.176 | −0.205 | −0.071 | 0.471 |
| [AgL3(N3,N3)]+ | 6c | −0.175 | −0.203 | −0.065 | 0.485 |
| [AgL3(N3,S)]+ | 6d | −0.181 | −0.203 | −0.059 | 0.563(0.478)b |
| [Ag2L3(N1,N1)2]2+ | 6e | −0.205 | −0.180 | −0.051 | 0.467 |
| [Ag2L3(N1,N1)2]2+ | 6f | −0.205 | −0.178 | −0.051 | 0.462 |
| [AgL3(N1)2]+ | 6g | −0.206 | −0.179 | −0.051 | 0.462 |
| [Ag2L3(N1,N1)L3(N1)2]2+ | 6h | −0.206 | −0.055 | −0.180 | 0.462 |
| L4 | 7a | −0.206(−0.199)b | −0.214(−0.207)b | −0.121 | 0.412(0.401)b |
| [AgL4(N1,N3)]+ | 7b | −0.204(−0.180)b | −0.182(−0.207)b | −0.095(−0.102)b | 0.477(0.486)b |
| [AgL4(N1,N3)]+ | 7c | −0.175(−0.203)b | −0.206(−0.187)b | −0.103(−0.95)b | 0.490(0.472)b |
| [AgL4(N3,N3)]+ | 7d | −0.176 | −0.204 | −0.104 | 0.485 |
| [AgL4(N1,N1)]+ | 7e | −0.204 | −0.174(−0.181)b | −0.097 | 0.475 |
| L5a | 8a | −0.198(−0.202)b | −0.209(−0.213)b | −0.102(−0.089)b | 0.419(0.409)b |
| [AgL5a(N3,N3)]+ | 8b | −0.178 | −0.202 | −0.082 | 0.475 |
| [AgL5a(S,S)]+ | 8c | −0.183(−0.178)b | −0.192(−0.167)b | 0.075(−0.052)b | 0.560(0.505)b |
| [AgL5a(S,N3,S)]+ | 8d | −0.167(−0.179)b | −0.194 | −0.066 | 0.531(0.575)b |
| [Ag2L5a(N1,N1)2]2 | 8e | −0.198 | −0.177 | −0.058 | 0.468 |
| [Ag2L5a(N1,N1)2]2+ | 8f | −0.185(−0.199)b | −0.162(−0.179)b | −0.062(−0.058)b | 0.476 |
The presence of bulky substitutes adjacent to the N1 and N3 nitrogen atoms can also influence the coordination selectivity revealed by either of these donor centers. Obviously, the S–CH3 groups influence the accessibility of the N1 and N3 donor atoms during the binding process identically. The above mentioned almost equal energy values for cis- and trans-conformers for the ligand L1 testify to this fact also. The determining contribution supposedly belongs to the more bulky N4-alkyl substitutes preventing the N3-coordination of the metal ion.
Conclusions
By applying the competitive extracting experiment we have demonstrated that the investigated pyrimidinophanes and their acyclic counterparts are capable of the selective binding of the Ag+ ions in the presence of other metals (Na+, Ca+, Co2+, Ni+, Cu2+, Zn2+, Cd2+). According to the X-ray analysis, the metal cation in AgL12 complexes is coordinated only by means of the N1 atoms of two 2-thiocytosine molecules. In all the complexes investigated by the X-ray method, these molecules are associated with the adjacent complex molecules through π⋯π stacking interactions.NMR diffusion experiments (CDCl3/DMSO) have demonstrated that podand L3 and pyrimidinophanes L5a,b actually form polynuclear and dimer Ag+ complexes with “an effective” 1:1 complex stoichiometry. Only the podand L4 forms mononuclear Ag+ complexes. The varying a solvent as well as the applying NO3− counter-ion instead of Pic− drastically affect the supramolecular structure of the silver complexes of L4 in the solution, leading to the formation of the stable in time gel system.
The combined analysis of NMR data and DFT calculations was successfully applied for the estimation of the most reliable complex structures. The analysis of the data obtained made it possible for us to conclude that for all the investigated pyrimidinophanes as well as for their acyclic counterparts the coordination of the Ag+ ion is brought about by means of the N1 nitrogen atoms of 2-thiocytosine fragments. The presence of a polymethylene spacer between the amine groups in the investigated pyrimidines makes unfavourable the N1,N1-coordination of the Ag+ ion by one molecule of the podand L3 or pyrimidinophanes L5a,5b and leads to the formation of duplex-type multi-component complexes. In fact, these results represent substantial progress in the study of receptor–structure relationships and could be the object for a further design of compounds of this type.
Experimental section
General remarks
All reagents and solvents were obtained from commercial sources. DMSO-d6 (99.5% isotopic purity) and CDCl3 (99.8% isotopic purity) for NMR spectroscopy was used from Aldrich.Microanalyses of C, H and N were carried out with a CHN-3 analyzer. Melting points of compounds were measured with a Boetius hotstage apparatus. The IR absorption spectra were recorded in KBr pellets on a Vector-22 FT-IR spectrometer (Bruker) with a resolution of 4 cm−1 and 16 scan accumulation. The NMR spectra were detected on a Bruker AVANCE-600 spectrometer equipped with a gradient block (field gradient to 50 g cm−1); working frequency of the spectrometer being 600.13 MHz in 1H, 150.90 MHz in 13C and 60.81 MHz in 15N experiments. A 5 mm inverse broadband probehead with the gradient coil along the Z axis was used. The spectra were recorded at 303 ± 0.1 K. The chemical shifts are presented relatively to Me4Si as the internal standard for samples in CDCl3 and signals of the residual protons of DMSO for samples in CDCl3-DMSO-d6 (δ(DMSO) = 2.50 ppm).
Synthesis of Ag+ complexes with ligand L1
Competitive extraction of metal cations
The CHCl3 was saturated with H2O to prevent volume changes during the extraction. Aqueous solution (10 ml) of an equimolar mixture of metal salts (NaNO3; Ca(NO3)2·4H2O, Co(NO3)2·6H2O, Ni(NO3)2·6H2O, Cu2(CH3COO)4·2H2O, Zn(NO3)2·4H2O, AgNO3 and Cd(CH3COO)2·2H2O with concentrations 1 × 10−4 M each) and HPic (2 × 10−4 M) was buffered at pH 7.3. Tris(hydroxymethyl)aminomethane–HCl (0.05 M) was used as a buffer. The aqueous mixture of metal–picrate salts and CHCl3 solution of extractant (10 ml, CL1 = 4 × 10−4 M and CL3,L4,L5a,L5b = 2 × 10−4 M) were magnetically stirred in a flask. The extraction equilibrium was reached after vigorous stirring for 1.5 h at 25 °C. Then two phases were allowed to settle for 1 h and afterwards separated by centrifugation. The relative concentrations of the cations in the aqueous phase were determined by applying of the atomic absorbance spectrometer AAS 1N (Carl Zeiss Jena) with the use of oxidative air-acetylene flame. Quantification was made by referring on a standard solution containing a mixture of salts. Blank experiments without added hosts were carried out under the same experimental conditions.NMR experiments on measuring the self-diffusion coefficient
The Fourier transform pulsed-gradient spin-echo (FT PGSE) experiments18–21 were performed by BPP-STE-LED (bipolar pulse pair-stimulated echo-longitudinal Eddy current delay) sequence.22 Data were acquired with a 50.0 ms diffusion delay, with bipolar gradient pulse duration from 2.8 to 4.0 ms (depending on the system under investigation), 1.1 ms spoil gradient pulse (30%) and a 5.0 ms eddy current delay. The bipolar pulse gradient strength was varied incrementally from 0.01 to 0.32 T m−1 in 16 steps.The diffusion experiments were performed at least three times and only the data with the correlation coefficients of a natural logarithm of the normalized signal attenuation (lnI/I0) as a function of the gradient amplitude b = γ2δ2g2(Δ − δ/3) (γ is the gyromagnetic ratio, g is the pulsed gradient strength, Δ is the time separation between the pulsed-gradients, δ is the duration of the pulse) higher than 0.999 were included. The standard deviations of the self-diffusion coefficients determination did not exceed 5%.
The volume changes of free and complexed ligands were estimated from the simple ratio:
| Vcomp/Vfree = (DH,free/DH,obs)3 |
X-ray crystallographic study
The crystals of Ag·L12·Pic,12 Ag·L12·NO3 and Ag2·L12·(NO3)2 complexes were obtained by evaporation from their saturated MeOH solutions. The single-crystal data for all compounds were collected on a Smart Apex II automatic diffractometer using graphite monochromated radiation. The structures were solved by direct methods and refined by full-matrix least-squares using the SHELXL97 program.23 All the non-hydrogen atoms were refined with anisotropic atomic displacement parameters. Hydrogen atoms were placed in the calculated positions. All figures were made using the program Mercury.†24Crystallographic data for the complex Ag·L12·Pic (AgC22H28N9O7S2) at 150(2) K: M = 702.52, triclinic system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.032 (1) Å, b = 10.150 (1) Å, c = 15.784 (2) Å, α = 100.504 (1), β = 94.365 (1), γ = 117.014 (1)°, Z = 2, V = 1384.5 (3) Å3, ρcalc = 1.685 g cm−3, μ(MoKα) = 0.71073 Å, crystal dimensions of 0.10 × 0.20 × 0.30 mm. 5374 independent reflections (Θmax = 26.00°) and 377 parameters, R1 = 0.0334 and wR2 = 0.0864.
, a = 10.032 (1) Å, b = 10.150 (1) Å, c = 15.784 (2) Å, α = 100.504 (1), β = 94.365 (1), γ = 117.014 (1)°, Z = 2, V = 1384.5 (3) Å3, ρcalc = 1.685 g cm−3, μ(MoKα) = 0.71073 Å, crystal dimensions of 0.10 × 0.20 × 0.30 mm. 5374 independent reflections (Θmax = 26.00°) and 377 parameters, R1 = 0.0334 and wR2 = 0.0864.
Crystallographic data for the complex Ag·L12·NO3 (AgC16H26N7O3S2) at 296(2) K: M = 536.43, triclinic system, space group P, a = 8.112 (2) Å, b = 9.971 (2) Å, c = 15.157 (3) Å, α = 96.068 (4), β = 90.565 (5), γ = 99.987 (5)°, Z = 2, V = 1200.1 (4) Å3, ρcalc = 1.485 g cm−3, μ(MoKα) = 0.71073 Å, crystal dimensions of 0.11 × 0.12 × 0.15 mm, 4648 independent reflections (Θmax = 26.00°) and 256 parameters, R1 = 0.0868 and wR2 = 0.3054.
Crystallographic data for dimer complex Ag2·L12·(NO3)2 (Ag2C16H26N8O6S2) at 150(2) K: M = 706.31, monoclinic system, space group P21/n, a = 10.094 (3) Å, b = 20.188 (6) Å, c = 12.319(4) Å, β = 99.246 (3)°, Z = 4, V = 2477.8 (12) Å3, ρcalc = 1.893 g cm−3, μ(MoKα) = 0.71073 Å, crystal dimensions of 0.10 × 0.15 × 0.20 mm, 4862 independent reflections (Θmax = 26.00°) and 308 parameters R1 = 0.0321 and wR2 = 0.0904.
Theoretical methods
Quantum-chemical calculations were carried out using the Orca 2.9 program package25 and employing the density functional theory (DFT), applying GGA-type PBE functional.26 Atomic orbitals were described using the contracted TZVP basis set reported by Ahlrichs et al.27,28All computations were performed with full optimization of the molecular geometry without any symmetry constrains taking into account solvent effects within the COSMO model.29 This optimization was followed by calculations of vibrational spectra to ensure that the optimized geometries correspond to minima on the total potential energy surfaces (no imaginary frequencies). These calculations were also used to evaluate the thermal and vibrational corrections to the enthalpies (at 298.15 K and 1 atm). The effective atomic charges for optimized structures were obtained from the Löwdin population analysis.
The preorganization energy of the ligand ΔEpreorg was calculated as:
| ΔEpreorg = Ecomplexed(L) − EL | (10) |
For the optimized ligand structures and their complexes, a special procedure30 for calculation of chemical shifts in NMR spectra of these compounds was used. The choice of an optimal model of the complex has been accomplished using a deviation of the calculated and experimental values of the chemical shift differences Δδ1H and Δδ15N for free ligand and its complex. Nuclei 13C appeared to be not quite perceptible towards the complex formation with silver ions. The experimental values Δδ13C did not exceed 1.5 ppm.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (grants 13-03-00709 and 15-43-02130).Notes and references
- H. Yang, K. L. Metera and H. F. Sleiman, Coord. Chem. Rev., 2010, 254, 2403–2415 CrossRef CAS PubMed.
- G. H. Clever and M. Shionoya, Coord. Chem. Rev., 2010, 254, 2391–2402 CrossRef CAS PubMed.
- I. Okamoto, T. Ono, R. Sameshima and A. Ono, Chem. Commun., 2012, 48, 4347–4349 RSC.
- A. Ono, H. Togiroe, Y. Tanaka and I. Okamoto, Chem. Soc. Rev., 2011, 40, 5855–5866 RSC.
- M. A. Galindo, D. Amantia, W. Clegg, R. W. Harrington, R. J. Eyre, J. P. Goss, P. R. Briddon, W. McFarlane and A. Houlton, Chem. Commun., 2009, 45, 2833–2835 RSC.
- J. L. Sessler, C. M. Lawrence and J. Jayawickramarajah, Chem. Soc. Rev., 2007, 36, 314–325 RSC.
- B. Lippert, Coord. Chem. Rev., 2000, 200–202, 487–516 CrossRef CAS.
- J. A. R. Navarro and B. Lippert, Coord. Chem. Rev., 2001, 222, 219–250 CrossRef CAS.
- J. J. Fiol, M. Barceló-Oliver, A. Tasada, A. Frontera, À. Terrón and À. García-Raso, Coord. Chem. Rev., 2013, 257, 2705–2715 CrossRef CAS PubMed.
- M. Goodgame and D. A. Jakwbovic, Coord. Chem. Rev., 1987, 79, 97–134 CrossRef CAS.
- V. E. Semenov, A. S. Mikhailov, A. D. Voloshina, N. V. Kulik, A. D. Nikitashina, V. V. Zobov, S. V. Kharlamov, S. K. Latypov and V. S. Reznik, Eur. J. Med. Chem., 2011, 46, 4715–4724 CrossRef CAS PubMed.
- S. N. Podyachev, V. E. Semenov, V. V. Syakaev, N. E. Kashapova, S. N. Sudakova, J. K. Voronina, A. S. Mikhailov, A. D. Voloshina, V. S. Reznik and A. I. Konovalov, RSC Adv., 2014, 4, 10228–10239 RSC.
- P. Yakovchuk, E. Protozanova and M. D. Frank-Kamenetskii, Nucleic Acids Res., 2006, 34, 564–574 CrossRef CAS PubMed.
- P. S. Pregosin, P. G. A. Kumar and I. Fernández, Chem. Rev., 2005, 105, 2977 CrossRef CAS PubMed.
- I. Fernandez and P. S. Pregosin, Magn. Reson. Chem., 2006, 44, 76–82 CrossRef CAS PubMed.
- A. Yiu-Yan Tam and V. Wing-Wah Yam, Chem. Soc. Rev., 2013, 42, 1540–1567 RSC.
- D. Braga, S. d'Agostino, E. D'Amen, F. Grepioni, D. Genovese, L. Prodi and M. Sgarzi, Dalton Trans., 2013, 42, 16949–16960 RSC.
- P. Stilbs, Prog. Nucl. Magn. Reson. Spectrosc., 1987, 19, 1 CrossRef CAS.
- Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed. Engl., 2005, 44, 520 CrossRef CAS PubMed.
- T. Brand, E. J. Cabrita and S. Berger, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 46, 159 CrossRef CAS PubMed.
- P. S. Pregosin, P. G. A. Kumar and I. Fernández, Chem. Rev., 2005, 105, 2977 CrossRef CAS PubMed.
- D. Wu, A. Chen and C. S. Johnson, J. Magn. Reson., Ser. A, 1995, 115, 260–264 CrossRef CAS.
- G. M. Sheldrick, SHELX-97. Programs for Crystal Structure Analysis (Release 97-2), University of Göttingen, Germany, 1997 Search PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed.
- Orca. An ab initio, DFT and semiempirical SCF-MO package, 2012 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3855–3868 CrossRef.
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef PubMed.
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef PubMed.
- S. Sinnecker, A. Rajendran, A. Klamt, M. Diedenhofen and F. Neese, J. Phys. Chem. A, 2006, 110, 2235–2245 CrossRef CAS PubMed.
- Calculation of NMR and EPR Parameters. Theory and Applications, ed. M. Kaupp, M. Bühl and V. Malkin, Wiley-VCH, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Calculated enthalpy and entropy values for the formation of the complex isomers, the coordinates for all geometry-optimized structures. CCDC 1012021–1012023. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra14070b |
| This journal is © The Royal Society of Chemistry 2015 |