
Ni-enhanced Co3O4 nanoarrays grown in situ on a Cu substrate as integrated anode materials for high-performance Li-ion batteries†
Abstract
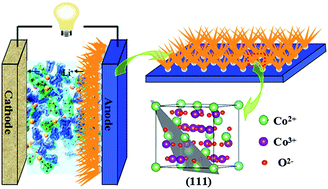
A two-step strategic approach was proposed to synthesize three-dimensional Co3O4 nanoarrays fabricated on a Cu substrate surface with a Ni layer as the interface. Firstly, a Ni-nanoseed-layer was prepared on a Cu substrate by electrodepositing Ni. And then Co3O4 nanoarrays were in situ grown on the Ni layer via a hydrothermal synthesis. The as-obtained materials were used directly as anodes for Li-ion batteries, and the electrodes maintained a high capacity up to 1150 mA h g−1 at 0.1 C after 30 cycles, and showed an excellent cycling stability and rate capability. The good electrochemical performance is owing to the pre-electrodeposited Ni-nanoseed-layer, because the Ni layer could improve the mechanical adhesion between Co3O4 nanoarrays and substrates effectively and increase the conductivity of anodes, without applying binders or conductive additives. This strategic in situ synthesis method will probably open a new avenue for the development of integrated electrode materials for high-performance Li-ion batteries.
 Please wait while we load your content...
Please wait while we load your content...

