
A supramolecular recyclable catalyst for aqueous Suzuki–Miyaura coupling†
Miao Qiab,
Pei Zi Tanab,
Fei Xueab,
Haripal Singh Malhiac,
Zhong-Xing Zhang*a,
David J. Young*acd and
T. S. Andy Hor*ab
aInstitute of Materials Research and Engineering, A*STAR (Agency for Science, Technology and Research), 3 Research Link, Singapore 117602, Singapore. E-mail: andyhor@imre.a-star.edu.sg; zhangzx@imre.a-star.edu.sg; Tel: +65-68744341
bDepartment of Chemistry, Faculty of Science, National University of Singapore, 3 Science, Drive 3, 117543, Singapore. E-mail: andyhor@nus.edu.sg
cFaculty of Science, Universiti Brunei Darussalam, Jalan Tungku Link, Gadong BE1410, Brunei Darussalam
dSchool of Science, Monash University Malaysia, 47500 Bandar Sunway, Selangor D.E., Malaysia. E-mail: david.james.young@monash.edu; Tel: +603 5514 5674
First published on 2nd December 2014
Abstract
A water-soluble, supramolecular catalytic system has been designed based on inclusion complexation between a hydrophobic palladium(II)–dipyrazole complex bearing an adamantyl (Ad) molecular recognition moiety and a complementary, hydrophilic β-cyclodextrin (β-CD) derivative. The single-crystal molecular structure of the Pd(II) complex was determined and its host–guest inclusion complexation with heptakis(2,6-di-O-methyl)-β-CD (dmβ-CD) in an aqueous medium was confirmed by 2D NOESY 1H NMR spectroscopy. The catalyst showed high activity for Suzuki–Miyaura coupling of hydrophilic aryl bromides with aryl boronic acids in aqueous organic solvents. In the presence of n-Bu4NBr as a stabilizer, the catalyst-containing reaction solution can be recycled and reused multiple times to catalyze the coupling reaction of fresh substrates once the product has been removed by centrifugation. This work demonstrates a supramolecular complex approach, non-covalently modifying a water insoluble metal complex to provide a water-soluble inclusion system to serve as a recyclable catalyst for potential application in green chemical synthesis.
Introduction
Green chemistry addresses the environmental impact of both chemical products and the processes by which they are produced.1–4 Water is an ideal green solvent and may offer additional benefits such as improved reactivities, selectivities, simplified workup procedures and recycling of an expensive metal catalyst.5,6The development of novel water soluble catalysts has elicited interest from both industrial and academic researchers.7,8 Designing catalytically active metal species to operate in an aqueous phase has primarily focused on ligands with suitable hydrophilic groups in addition to their functionality necessary to support the stability, activity, and selectivity of the metal center. Most commonly, ionic substituents such as sulfonate, carboxylate, phosphonate, or ammonium are employed, but neutral hydrophilic polyols, carbohydrates, and polyethylene glycols have also been successfully used.9
We herein present a new approach to the design and preparation of water-soluble metal catalysts by utilizing non-covalent rather than covalent bonding. This strategy makes use of host–guest inclusion complexation to reversibly tether a hydrophilic moiety to the hydrophobic ligand of the metal complex thereby enhancing its solubility in aqueous media.
Cyclodextrins (CDs) are well-known cyclic oligosaccharides formed from the enzymatic degradation of starch.10 Their partial water solubility, homochirality and ability to form inclusion complexes makes them among the most studied hosts in supramolecular chemistry,11–16 with a wide range of applications from drug delivery,17–19 to resolution of enantiomers20 and as catalysts.21–23 The supramolecular structures formed between CDs and polymers have also inspired interesting supramolecular biomaterials. Some of us have recently constructed a series of supramolecular hydrogels, non-covalently connected micelles, nano-capsules and pseudo-block copolymers based on the inclusion complexation between β-CD moieties and adamantyl (Ad) groups.24–27
On the basis of this experience with the self-assembling behavior of β-CD/Ad pairs and of transition metal complexes bearing pyrazole-derived ligands,28 we have now designed a bidentate dipyrazole ligand tethered to an Ad group as the molecular recognition moiety (Ad-L). This ligand then reacted with the Pd(II) precursor PdCl2(NCCH3)2 to form an Ad-containing complex (Ad-L–PdCl2) that is insoluble in water because of the bulky hydrophobic group. Upon host–guest inclusion complexation of Ad-L–PdCl2 with heptakis(2,6-di-O-methyl)-β-CD (dmβ-CD) in aqueous media, however, the Pd(II) complex dissolves completely at room temperature to form a water soluble supramolecular assembly (Ad-L–PdCl2 ⊂ β-CD) that efficiently catalyzes Suzuki–Miyaura coupling between hydrophilic aryl bromides with aryl boronic acid at room temperature. By comparison, the Pd(II) complex (Ad-L–PdCl2), that is present heterogeneously in the aqueous reaction mixture exhibits much lower catalytic efficiency under similar conditions.
Results and discussion
Synthesis and characterization of Ad-L and Ad-L–PdCl2
The ligand Ad-L was synthesized by the nucleophilic substitution reaction of one equivalent of 1-adamantanemethylamine with two equivalents of (3,5-dimethyl-1H-pyrazolyl)methanol in anhydrous acetonitrile at room temperature over 24 h (Scheme 1). This facile procedure resulted in good yields with little side products. The central amine and the two pyrazolyl nitrogen atoms of this ligand are potential ligand donors to the metal centre and hence this hybrid molecule can possibly bind to a metal in a κ2(N,N) or κ3(N,N′,N) fashion depending on the electronic state and steric constraints around the metal.29 The co-presence of a tertiary amine and pyrazolyl nitrogen donors in proximity also enhances the hemilability which gives it coordinative freedom and conformational freedom to adapt to the coordination and geometry changes of the metal during the catalytic cycle.30,31 | ||
| Scheme 1 Synthesis of Ad-L and Ad-L–PdCl2. | ||
The adamantyl-containing Pd(II) complex (Ad-L–PdCl2) was quantitatively prepared by reacting Ad-L with an equal amount of PdCl2(NCCH3)2 in dry CH2Cl2 at room temperature. The 1H NMR spectrum of the ligand showed a 0.1–0.4 ppm downfield shift upon coordination. Most affected was the –CH2– group bridging the central amine and the pyrazolyl group. From a singlet in the free ligand, this signal became two doublets at 7.11 and 5.23 ppm with a geminal coupling constant of 15 Hz (see ESI, Fig. S1†).
Single crystals of adamantyl-containing Pd(II) complex suitable for single-crystal X-ray diffraction analysis (Fig. 1) were obtained by slow diffusion of diethyl ether into a solution of complex in CH2Cl2 at −20 °C. Selected bond lengths and bond angles are summarized in Table 1.
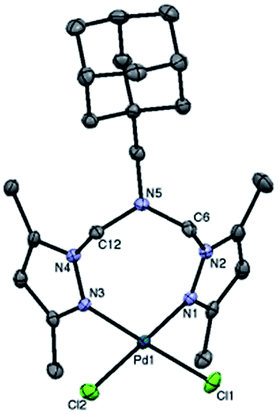 | ||
| Fig. 1 ORTEP diagram of adamantyl-containing Pd(II) complex shown with 50% probability ellipsoids (hydrogen atoms are omitted for clarity). | ||
| Bond length (Å) | Bond angle (°) |
|---|---|
| Pd(1)–N(3) 2.019(2) | N(3)–Pd(1)–N(1) 88.44(8) |
| Pd(1)–N(1) 2.024(2) | N(3)–Pd(1)–Cl(1) 177.60(6) |
| Pd(1)–Cl(1) 2.2777(6) | N(1)–Pd(1)–Cl(1) 89.20(6) |
| Pd(1)–Cl(2) 2.2858(6) | N(3)–Pd(1)–Cl(2) 90.01(6) |
| N(1)–Pd(1)–Cl(2) 177.62(6) | |
| Cl(1)–Pd(1)–Cl(2) 92.33(3) |
The Pd(II) centre in Ad-L–PdCl2 is slightly distorted from the ideal square planar geometry that is typically found in a d8 metal centre. The bidentate chelating ligand binds the Pd(II) centre through the two pyrazolyl nitrogen atoms N3 and N1. The non-coordinating [N5⋯Pd] distance is 3.683 Å. This κ2(N,N) coordination gives rise to a puckered 8-membered metallocyclic ring –Pd1–N1–N2–C6–N5–C12–N4–N3– which is best described as a boat–chair like conformation. The Pd–N (pyrazolyl) bond lengths are 2.019(2) Å and 2.024(2) Å which are consistent with literature values32–34 as are the Pd–Cl distances of 2.2777(6) Å and 2.2858(6) Å.32–35
Host–guest inclusion complexation between Ad-L–PdCl2 and β-CD derivative
The adamantyl (Ad) group and β-CD moiety is a well-known host–guest pair with a stability constant (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K) up to 5 at room temperature.36–38 To confirm the host–guest inclusion complexation of the adamantyl-containing Pd(II) complex with heptakis(2,6-di-O-methyl)-β-CD (dmβ-CD), 2D-NOESY 1H NMR measurements were made in aqueous DMSO. Dmβ-CD was chosen for this analysis because of its enhanced solubility in water relative to β-CD.25
K) up to 5 at room temperature.36–38 To confirm the host–guest inclusion complexation of the adamantyl-containing Pd(II) complex with heptakis(2,6-di-O-methyl)-β-CD (dmβ-CD), 2D-NOESY 1H NMR measurements were made in aqueous DMSO. Dmβ-CD was chosen for this analysis because of its enhanced solubility in water relative to β-CD.25
The sample was prepared by adding a solution of Ad-L–PdCl2 (guest) in DMSO-d6 into a solution of dmβ-CD (host) in D2O. The final host–guest mixture (molar ratio 1:1) in D2O–DMSO-d6 (1:1, v/v) was filtered before measurement. It can be seen in the 2D NOESY spectrum (Fig. 2, left) that the peaks of the Pd(II) complex were only present at low intensity compared to the peaks belonging to dmβ-CD. This is most likely due to longer proton relaxation times and the high hydrophobicity of the former. This is a common phenomenon for amphiphilic materials like copolymers in aqueous solution.26 The C(3)–H and C(5)–H of dmβ-CD (3.6 to 3.7 ppm) point into the hydrophobic cavity of β-CD and exhibit correlation signals with the protons (1.6 to 2.0 ppm) on the guest Ad group (Fig. 2, right), indicating that the Ad moiety has inserted into the β-CD cavity. This observation is in agreement with literature reports by us and by other groups.26,27,39
 | ||
| Fig. 2 (Left) 2D NOESY 1H NMR spectrum of Ad-L–PdCl2 and dmβ-CD in D2O–DMSO-d6 (1:1, v/v) with water suppression (500 MHz, 25 °C); (right) expanded 2D NOESY 1H NMR spectrum. | ||
Suzuki–Miyaura coupling reaction
In this work, Suzuki–Miyaura cross coupling of 4-bromobenzoic acid and phenylboronic acid was used to evaluate the catalytic performance of our supramolecular catalyst (Ad-L–PdCl2 ⊂ dmβ-CD) (Scheme 2) in a series of water–organic solvent mixtures (3/1, v/v). These reactions were carried out in air at room temperature with a 0.5 mol% catalyst loading. The supramolecular pre-catalyst and the substrates dissolved in the water–organic solvent reaction media to form a homogeneous reaction mixture. Product yields after 2 hours are given in Table 2.
 | ||
| Scheme 2 Suzuki–Miyaura coupling reaction of 4-bromobenzoic acid and phenylboronic acid in aqueous media catalyzed by supramolecular inclusion complex catalyst (Ad-L–PdCl2 ⊂ dmβ-CD). | ||
| Entry | Reaction media | Yieldb (%) |
|---|---|---|
| a Reaction conditions: 0.5 mol% supramolecular catalyst (Ad-L–PdCl2 complexed with an equal amount of dmβ-CD), 0.5 mmol of 4-bromobenzoic acid, 0.6 mmol of phenylboronic acid, 1.2 mmol of Na2CO3, 4 mL of water–organic mix solvent (3/1, v/v), room temperature for 2 h in air.b Yields were determined by 1H NMR analysis in d6-acetone using an internal standard (hexadecane) and calculated using a product calibration plot with R2 = 0.998 (see ESI). | ||
| 1 | H2O–CH3OH | >97 |
| 2 | H2O–DMF | 87.7 |
| 3 | H2O–CH3CN | 31.3 |
| 4 | H2O–acetone | 61.2 |
It can be seen from Table 2 that the yield of the Suzuki–Miyaura coupling product after 2 hours decreased in the order H2O–CH3OH > H2O–DMF > H2O–acetone > H2O–CH3CN. Fortuitously, the H2O–CH3OH mixture is also the ‘greenest’ of these water–organic solvent mixtures.49
 | ||
| Fig. 3 (Top) Comparison of Suzuki–Miyaura coupling reactions performed in H2O–CH3OH (3/1, v/v) with and without dmβ-CD in air at room temperature over 4 h. Each point represents the mean value ± SD of 2 runs; (bottom) photographs comparing the homogeneity of catalyst and catalyst mixed with 4-bromobenzoic acid/Na2CO3 in H2O–CH3OH (3/1, v/v), as well as the turbidity of the reaction mixtures after adding phenylboronic acid in 30 min with (a1–a3) and without (b1–b3) the host dmβ-CD, respectively. | ||
The influence of solubility was further investigated by varying the proportion of organic solvent in the presence and absence of the host dmβ-CD (Fig. 4). The reactions in the presence of dmβ-CD always afforded the product in higher yields (94–99%) over a volume ratio of H2O–CH3OH ranging from 0.5 to 5. The reactions in the absence of dmβ-CD also afforded the product in good yields (94–98%), but only when the volume ratio of H2O–CH3OH was below 3. Interestingly, when the volume ratio of H2O–CH3OH was higher than 3, the yields dropped to around 50% because of poor aqueous solubility of the catalyst. When the volume ratio of H2O–CH3OH was in the range 0.5–2, the Pd(II) complex either dissolved or was well dispersed in the reaction medium, even in the absence of dmβ-CD. Therefore the yields in both cases (i.e. with and without dmβ-CD) were similar. When the volume ratio of H2O–CH3OH was 3, the Pd(II) complex was kept soluble in the reaction medium by the dmβ-CD. However, when dmβ-CD was absent, the Pd(II) complex dissolved poorly resulting in a lower yield (around 86%) and this was exacerbated by increasing the volume ratio of H2O–CH3OH to 4 and 5 (yield around 50%).
 | ||
| Fig. 4 Comparison of Suzuki–Miyaura coupling performed in various proportions of H2O and CH3OH, with and without dmβ-CD (conditions: room temperature, 2 h in air). Each point represents the mean value ± SD of 2 runs. | ||
Recycling of the catalyst
High precious metal prices and toxicity make recycling of the Pd catalyst attractive from an economic and environmental perspective. Recycling experiments were performed using the standard Suzuki–Miyaura coupling reaction of 4-bromobenzoic acid and phenylboronic acid in water–MeOH (3:1, v/v). The coupling product was easily isolated by centrifugation. The aqueous filtrate was recovered and investigated for its ability to catalyze the reaction of freshly added substrates (Table 3).
| Entry | Stabilizer | Stabilizer/aryl bromide (mol/mol) | Yield for each recycling stepb (%) | ||||
|---|---|---|---|---|---|---|---|
| Step 0 | Step 1 | Step 2 | Step 3 | Step 4 | |||
| a Reaction conditions: 4-bromobenzoic acid (1.0 equiv., 0.125 mol L−1), phenylboronic acid (1.2 equiv.), initial catalyst loading (0.5 mol%, for step 0), pH (11.0, at the beginning of each step), room temperature for 2 h in air in water–MeOH (3:1, v/v).b Yields were determined by 1H NMR analysis in d6-acetone using an internal standard (hexadecane) and calculated using a product calibration plot with R2 = 0.998 (see ESI). |
|||||||
| 1 | — | — | 100 | 59.4 | 21.9 | 15.1 | 9.0 |
| 2 | TBAB | 0.1 | 100 | 62.4 | 32.8 | 21.4 | 18.8 |
| 3 | TBAB | 0.15 | 83.0 | 100 | 77.5 | 53.0 | 20.6 |
| 4 | TBAB | 0.20 | 69.2 | 92.2 | 69.7 | 52.4 | 15.6 |
| 5 | TBAB | 0.25 | 69.7 | 81.1 | 49.8 | 35.0 | 15.5 |
| 6 | TBAB | 1.0 | 8.4 | 25.6 | 15.4 | 18.7 | 14.6 |
| 7 | NaBr | 1.0 | 100 | 43.0 | 19.0 | 11.8 | 11.5 |
It has been reported that pH strongly influences catalyst activity for Suzuki–Miyaura cross coupling reactions conducted in aqueous media50 and that a pH of around 11 is optimal.50,51
In this work, the pH value of the original reaction mixture was measured to be ca. 11.0 at the beginning of the coupling reaction. However, by the end of the reaction, the pH value of the recycled filtrate dropped to be ca. 10.0 and then ca. 9.0 after the second reaction. Therefore, the pH of the reaction mixture was adjusted back to 11.0 with dilute aqueous NaOH before each recycling experiment.
The Suzuki–Miyaura coupling reaction of 4-bromobenzoic acid and phenylboronic acid in water–MeOH was quantitative after 2 h (Table 3, entry 1). However, this high catalytic activity was not maintained using the recycled catalyst. The yield dropped to ca. 60% at the first recycle (i.e. step 1) and was less than 10% by the 4th recycling step. Palladium black was observed in these reactions, thus suggesting catalyst decomposition.
Pd(0) intermediates are the catalytically active species in Suzuki–Miyaura cross coupling reactions.52–54 Catalyst decomposition to form palladium black is also frequently observed. This phenomenon is usually explained by assuming that the oxidative addition step which starts the catalytic cycle is in competition with an ill-defined sequence of reactions leading to palladium nanoparticles (Pd-NPs) that eventually aggregate irreversibly to form larger, catalytically inactive, palladium metal particles (palladium black).55 Obviously a low conversion will result if the aggregation is faster than the cross coupling. Catalyst decomposition also hampers recycling. A simple and effective approach to prevent the catalyst from losing activity is to use quaternary ammonium salts which stabilize Pd-NPs and slow the aggregation process.56–58 Stabilization of Pd-NPs can increase the catalytic activity because dispersible Pd-NPs are themselves active catalysts57 or act as a reservoir of soluble, catalytically active Pd(0).59,60 Both the cation and anion of ammonium salt play a combined role in stabilizing Pd-NPs, the anion being coordinated to the electrophilic metal nanoparticles with the cation surrounding the negatively-charged anion-metal sphere.55
In our work, a suitable amount of tetrabutylammonium bromide (TBAB) was found to be effective to stabilize the catalyst against decomposition and made recycling feasible. Experiments carried out using different TBAB–4-bromobenzoic acid molar ratios showed that the catalytic performance for each recycling step increased significantly with increasing the stabilizer until a maximum was reached at the TBAB–4-bromobenzoic acid molar ratio of 0.15:1 (Table 3, entries 2–6). When higher amounts of stabilizer were used, the substrate conversion decreased. When TBAB in recycling reaction (1) was increased from 0 to 0.15 equiv., the yield increased from ca. 60% (entry 1) to 100% (entry 3). When the TBAB was further increased to 0.25 equiv., the yield decreased from 100% to ca. 80% (entry 5) and to ca. 25% when 1.0 equivalents of TBAB were employed (entry 6). No catalyst decomposition was observed in these TBAB stabilized reactions. These results indicate that the optimal molar ratio of TBAB to substrate was 0.15 and that high loadings of TBAB actually inhibited the reaction, presumably at the oxidative addition step. By contrast, NaBr did not stabilize the catalyst and slight inhibition was observed (entry 7).
Although 0.15 equiv. of TBAB significantly enhanced the catalytic performance in subsequent recycling steps (Table 3, entry 3, step 1–4), this was not the case for the initial run (Table 3, entry 3, step 0). The yield for this step was quantitative without stabilizer and approximately 80% with stabilizer. It is possible that the higher stability gained from the TBAB stabilization leads to longer catalytic induction period. Further, a series of catalytic reactions and recycling experiments was carried out for 5 h and 24 h, and compared with those performed at 2 h in the presence of 0.15 equiv. of TBAB (Fig. 5). At a reaction time of 5 h (red bars), the aqueous solution with supramolecular catalyst can be recycled and reused twice before the yield dropped to below 90% and three times for a reaction time of 24 h (blue bars).
 | ||
| Fig. 5 Variation of yield with increasing reaction time in catalyst recycling experiments; 4-bromobenzoic acid (1.0 equiv., 0.125 mol L−1), phenylboronic acid (1.2 equiv.), initial catalyst loading (0.5 mol%, for step 0), TBAB (0.15 equiv.), pH (11.0, at the beginning of each step), room temperature, air, water–methanol (3:1, v/v). Each point represents the mean value ± SD of 2 runs. | ||
Experimental
Synthesis of bis[(3,5-dimethyl-1H-pyrazolyl)methyl][(1-adamantyl)methyl]amine (Ad-L)
The synthesis of Ad-L was modified from a previous report by our group.28 (3,5-Dimethyl-1H-pyrazolyl)methanol (0.252 g, 2 mmol) was dissolved in anhydrous CH3CN (30 mL). 1-Adamantanemethylamine (0.166 g, 1 mmol) was added and the solution stirred in a closed vessel for 24 h at room temperature. The solvent was removed under reduced pressure and the resultant white solid re-dissolved in 10 mL of n-hexane, filtered and the clear solution cooled overnight at 4 °C, yielding white crystals (0.26 g, yield 68%). 1H NMR (300 MHz, CDCl3): 1.21 (s, 6H, Ad-CH2–), 1.54 (d, 3H, J = 16.3 Hz, Ad-CHH′–), 1.61 (d, 3H, J = 12.3 Hz, Ad-CHH’′–), 1.84 (s, 3H, Ad-CH), 2.19 (s, 6H, Pz-CH3), 2.27 (s, 6H, Pz-CH3), 2.37 (s, 2H, Ad-CH2–N–), 4.91 (s, 4H, Pz-CH2–N–), 5.79 (s, 2H, Pz-CH). 13C NMR (75.5 MHz, CDCl3): δ (ppm) 12.1 (Pz-CH3), 14.2 (Pz-CH3), 29.1 (Ad-CH2), 35.4 (Ad-C), 37.7 (Ad-CH2), 41.1 (Ad-CH), 62.2 (Ad-CH2–N–), 69.0 (N–CH2-Pz), 106.3 (Pz-CH), 140.2 (Pz-C), 147.9 (Pz-C). ESI-MS (in CH2Cl2, m/z (%)): [M + H]+ = 381 (100), [2M + Na]+ = 784 (30). Anal. calcd for C23H35N5: C, 72.40; H, 9.25; N, 18.35. Found: C, 72.35; H, 8.38; N, 18.12%.Synthesis of adamantyl-containing Pd(II) complex (Ad-L–PdCl2)
PdCl2(NCCH3)2 was synthesized following a literature procedure.61 A suspension of PdCl2 (1.00 g, 5.65 mmol) was heated under reflux in CH3CN (50 mL) with vigorous stirring for 10 h under N2. The resultant wine-red colored solution was cooled for 16 h at 4 °C and a yellow-orange solid was filtered, washed with Et2O and dried in vacuo at room temperature. Solutions of PdCl2(NCCH3)2 (0.070 g, 0.27 mmol) in dry CH2Cl2 (10 mL) and Ad-L (0.103 g, 0.27 mmol) in dry CH2Cl2 (5 mL) were mixed and stirred at room temperature for 12 h (ref. 62) and then concentrated under vacuum. Et2O (5 mL) was added to induce precipitation. A yellow-orange solid was obtained, washed with Et2O, and dried under vacuum. The product was crystallized from a CH2Cl2–Et2O mixture to give bright orange crystals (0.14 g, yield 92%). 1H NMR (500 MHz, CDCl3): 1.64 (s, 6H, Ad-CH2–), 1.70 (d, 3H, J = 18.9 Hz, Ad-CHH′–), 1.78 (d, 3H, J = 11.9 Hz, Ad-CHH′–), 2.07 (s, 3H, Ad-CH), 2.14 (s, 2H, Ad-CH2–N–), 2.36 (s, 6H, Pz-CH3), 2.78 (s, 6H, Pz-CH3), 5.23 (d, 2H, J = 15.1 Hz, Pz-CHH′–N–), 5.87 (s, 2H, Pz CH), 7.11 (d, 2H, J = 15.7 Hz, Pz-CHH′–N–). 13C NMR (125 MHz, CDCl3): δ (ppm) 13.6 (Pz CH3), 15.8 (Pz-CH3), 28.8 (Ad-CH2), 37.2 (Ad-C), 37.4 (Ad-CH2), 41.8 (Ad-CH), 57.6 (Ad-CH2–N–), 70.7 (N–CH2-Pz), 108.8 (Pz-CH), 143.7 (Pz-C), 151.9 (Pz-C). ESI-MS (in CH2Cl2, m/z (%)): [M − Cl]+ = 524 (100), [M + Na]+ = 576 (20). Anal. calcd for C23H35Cl2N5Pd: C, 49.43; H, 6.31; N, 12.53. Found: C, 48.66; H, 5.71; N, 11.91%.Measurements and experimental methods
General procedure for Suzuki–Miyaura coupling reaction
:1, v/v) with stirring at room temperature until a clear, pale yellow solution was formed. The catalyst solution and substrate solution were mixed together and stirred at room temperature. After the required time had elapsed, the reaction mixture was neutralized with dilute, aqueous HCl. The resulting white precipitate was filtered, washed with water and dried under vacuum at 50 °C.Yield calculation for Suzuki–Miyaura coupling reaction
Yields were calculated by 1H NMR spectroscopy based on the 4-bromobenzoic acid used using hexadecane as an internal standard. After the Suzuki–Miyaura coupling reaction, the reaction mixture was neutralized with dilute aqueous HCl and a measured amount of hexadecane (usually 14 mg) in ethyl acetate (4 mL) was added. The organic phase was separated, and the reaction mixture extracted with fresh ethyl acetate (3 × 3 mL). The organic extracts were combined, washed twice with water (5 mL), dried over anhydrous Na2SO4 and a 0.9 mL aliquot was filtered, evaporated at room temperature and re-dissolved in deuterated solvent (acetone-d6). The yield of the product was calculated from a calibration curve (Fig. S2,† R2 = 0.998).Catalyst recycling
After a standard Suzuki–Miyaura coupling reaction of 4-bromobenzoic acid and phenylboronic acid in water–methanol (3:1, v/v), the white coupling product was isolated by centrifugation (6000 rpm, r.t.). The clear filtrate was transferred to a clean vial, and measured amounts of fresh substrate were added with stirring at room temperature. The pH was adjusted to 11.0 using dilute aqueous NaOH. After the required time had elapsed, the white coupling product was isolated by centrifugation, and the clear filtrate was recycled and reused for the next coupling reaction.
Conclusions
A key catalytic design in this system is the incorporation of an adamantyl tether onto the ligand backbone that does not interfere with the catalytic process but activating its inclusion property to capture a β-cyclodextrin moiety and uses that to solubilise the entire supramolecular hybrid in an aqueous or aqueous–organic mixed media. The main challenge in this design is the chemical and thermal stability of the supramolecular hybrid catalysts and, very importantly, introduction of the inclusion pair (adamantyl and cyclodextrin here) must not infringe with the catalytic site thereby hurting the catalytic efficacy. The crystal and molecular structure of the adamantyl-tethered complex reported herein gives clear evidence that the bulky pendant is pointing away from the metal center and that its introduction does not interfere with the coordination sphere of the Pd(II). It also does not impose undesirable bonding of the amine with the metal, which has been witnessed in other functional hybrid ligand systems.28 Encouraged by these results, we are actively applying this simple yet powerful design to a range of other metal catalysts and catalytic reactions.Acknowledgements
This work was supported by EDB (Singapore)-GSK (GlaxoSmithKline) (Grant ID: R143-000492-592) and Institute of Materials Research and Engineering (IMRE), A*STAR (Agency for Science, Technology and Research), Singapore.Notes and references
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- G. Centi and S. Perathoner, Catal. Today, 2003, 77, 287–297 CrossRef CAS.
- P. T. Anastas, T. C. Williamson, D. Hjeresen and J. J. Breen, Environ. Sci. Technol., 1999, 33, 116A–119A CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- M.-O. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415–1427 RSC.
- C. J. Li, Chem. Rev., 2005, 105, 3095–3165 CrossRef CAS PubMed.
- V. Polshettiwar, A. Decottignies, C. Len and A. Fihri, ChemSusChem, 2010, 3, 502–522 CrossRef CAS PubMed.
- R. A. Sheldon, I. Arends, G. J. Ten Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774–781 CrossRef CAS PubMed.
- K. H. Shaughnessy, Chem. Rev., 2009, 109, 643–710 CrossRef CAS PubMed.
- J. Szejtli, Chem. Rev., 1998, 98, 1743–1753 CrossRef CAS PubMed.
- G. Wenz, B.-H. Han and A. Muller, Chem. Rev., 2006, 106, 782–817 CrossRef CAS PubMed.
- F. Hapiot, S. Tilloy and E. Monflier, Chem. Rev., 2006, 106, 767–781 CrossRef CAS PubMed.
- A. Douhal, Chem. Rev., 2004, 104, 1955–1976 CrossRef CAS PubMed.
- A. Harada, Acc. Chem. Res., 2001, 34, 456–464 CrossRef CAS PubMed.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1918 CrossRef CAS PubMed.
- S. A. Nepogodiev and J. F. Stoddart, Chem. Rev., 1998, 98, 1959–1976 CrossRef CAS PubMed.
- J. Li and X. J. Loh, Adv. Drug Delivery Rev., 2008, 60, 1000–1017 CrossRef CAS PubMed.
- M. E. Davis and M. E. Brewster, Nat. Rev. Drug Discovery, 2004, 3, 1023–1035 CrossRef CAS PubMed.
- K. Uekama, F. Hirayama and T. Irie, Chem. Rev., 1998, 98, 2045–2076 CrossRef CAS PubMed.
- D. W. Armstrong, T. J. Ward, R. D. Armstrong and T. E. Beesley, Science, 1986, 232, 1132–1135 CAS.
- B. L. Zhang and R. Breslow, J. Am. Chem. Soc., 1997, 119, 1676–1681 CrossRef CAS.
- M. T. Reetz and S. R. Waldvogel, Angew. Chem., Int. Ed. Engl., 1997, 36, 865–867 CrossRef CAS.
- E. Monflier, E. Blouet, Y. Barbaux and A. Mortreux, Angew. Chem., Int. Ed. Engl., 1994, 33, 2100–2102 CrossRef.
- Z.-X. Zhang, K. L. Liu and J. Li, Angew. Chem., Int. Ed., 2013, 52, 6180–6184 CrossRef CAS PubMed.
- J.-L. Zhu, K. L. Liu, Z. Zhang, X.-Z. Zhang and J. Li, Chem. Commun., 2011, 47, 12849–12851 RSC.
- Z.-X. Zhang, K. L. Liu and J. Li, Macromolecules, 2011, 44, 1182–1193 CrossRef CAS.
- Z.-X. Zhang, X. Liu, F. J. Xu, X. J. Loh, E.-T. Kang, K.-G. Neoh and J. Li, Macromolecules, 2008, 41, 5967–5970 CrossRef CAS.
- F. Xue, J. Zhao and T. S. A. Hor, Dalton Trans., 2011, 40, 8935–8940 RSC.
- R. Mathieu, G. Esquius, N. Lugan, J. Pons and J. Ros, Eur. J. Inorg. Chem., 2001, 2683–2688 CrossRef CAS.
- P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, 680–699 CrossRef CAS.
- Z. Weng, S. Teo and T. S. A. Hor, Acc. Chem. Res., 2007, 40, 676–684 CrossRef CAS PubMed.
- A. Boixassa, J. Pons, X. Solans, M. Font-Bardia and J. Ros, Inorg. Chim. Acta, 2003, 346, 151–157 CrossRef CAS.
- J. Garcia-Anton, J. Pons, X. Solans, M. Font-Bardia and J. Ros, Eur. J. Inorg. Chem., 2002, 3319–3327 CrossRef CAS.
- A. John, M. M. Shaikh, R. J. Butcher and P. Ghosh, Dalton Trans., 2010, 39, 7353–7363 RSC.
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1918 CrossRef CAS PubMed.
- M. Weickenmeier and G. Wenz, Macromol. Rapid Commun., 1996, 17, 731–736 CrossRef CAS.
- R. I. Gelb, L. M. Schwartz and D. A. Laufer, J. Chem. Soc., Perkin Trans. 2, 1984, 15–21 RSC.
- Y. Hasegawa, M. Miyauchi, Y. Takashima, H. Yamaguchi and A. Harada, Macromolecules, 2005, 38, 3724–3730 CrossRef CAS.
- T. Hanazawa, A. Koyama, T. Wada, E. Morishige, S. Okamoto and F. Sato, Org. Lett., 2003, 5, 523–525 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- F. Li and T. S. A. Hor, Adv. Synth. Catal., 2008, 350, 2391–2400 CrossRef CAS.
- S.-Q. Bai and T. S. A. Hor, Chem. Commun., 2008, 3172–3174 RSC.
- G. Herve, G. Sartori, G. Enderlin, G. Mackenzie and C. Len, RSC Adv., 2014, 4, 18558–18594 RSC.
- J. Tauchman, I. Cisarova and P. Stepnicka, Organometallics, 2009, 28, 3288–3302 CrossRef CAS.
- D. Dallinger and C. O. Kappe, Chem. Rev., 2007, 107, 2563–2591 CrossRef CAS PubMed.
- N. E. Leadbeater, Chem. Commun., 2005, 2881–2902 RSC.
- R. Franzen and Y. J. Xu, Can. J. Chem., 2005, 83, 266–272 CrossRef CAS.
- C. Capello, U. Fischer and K. Hungerbuhler, Green Chem., 2007, 9, 927–934 RSC.
- S. Ogo, Y. Takebe, K. Uehara, T. Yamazaki, H. Nakai, Y. Watanabe and S. Fukuzumi, Organometallics, 2006, 25, 331–338 CrossRef CAS.
- A. N. Marziale, D. Jantke, S. H. Faul, T. Reiner, E. Herdtweck and J. Eppinger, Green Chem., 2011, 13, 169–177 RSC.
- G. C. Fu, Acc. Chem. Res., 2008, 41, 1555–1564 CrossRef CAS PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- E. Amadio, A. Scrivanti, M. Bortoluzzi, M. Bertoldini, V. Beghetto, U. Matteoli and G. Chessa, Inorg. Chim. Acta, 2013, 405, 188–195 CrossRef CAS PubMed.
- H. Bonnemann, G. Braun, W. Brijoux, R. Brinkmann, A. S. Tilling, K. Seevogel and K. Siepen, J. Organomet. Chem., 1996, 520, 143–162 CrossRef.
- L. S. Ott and R. G. Finke, Coord. Chem. Rev., 2007, 251, 1075–1100 CrossRef CAS PubMed.
- M. T. Reetz and W. Helbig, J. Am. Chem. Soc., 1994, 116, 7401–7402 CrossRef CAS.
- V. P. Ananikov and I. P. Beletskaya, Organometallics, 2012, 31, 1595–1604 CrossRef CAS.
- I. W. Davies, L. Matty, D. L. Hughes and P. J. Reider, J. Am. Chem. Soc., 2001, 123, 10139–10140 CrossRef CAS.
- M. S. Kharasch, R. C. Seyler and F. R. Mayo, J. Am. Chem. Soc., 1938, 60, 882–884 CrossRef CAS.
- A. Panella, J. Pons, J. Garcia-Anton, X. Solans, M. Font-Bardia and J. Ros, Eur. J. Inorg. Chem., 2006, 1678–1685 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General measurement and experimental methods; a table of single crystal data and structure refinement parameters for adamantyl-containing Pd(II) complex (Ad-L–PdCl2); 1H NMR spectra of Ad-L and Ad-L–PdCl2; calibration curve based on 1H NMR analysis for the yield calculation for Suzuki–Miyaura coupling reaction. CCDC 1031688. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra13953d |
| This journal is © The Royal Society of Chemistry 2015 |