
A comprehensive examination of the self-disproportionation of enantiomers (SDE) of chiral amides via achiral, laboratory-routine, gravity-driven column chromatography†
Abstract
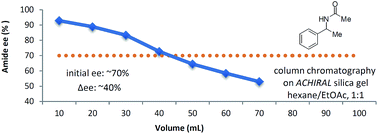
This work explores the self-disproportionation of enantiomers (SDE) of chiral amides via achiral, gravity-driven column chromatography as typically used in laboratory settings. The major findings of this work are: (1) the remarkable persistence and high magnitude of the SDE for the analytes under a variety of conditions, including polar solvents and different achiral stationary phases and (2) the notable generality of the SDE phenomenon as it occurs for a wide range of chiral amide substrates and even for a broad range of starting ee. This last aspect is unusual and not commonly observed. The key conclusion of this work is that it judiciously conveys the predictability of the SDE for chiral amides under the routine conditions of achiral chromatography. These results are consequently of concern for practitioners in the area of catalytic asymmetric synthesis involving chiral amides as intermediates or products and the inferents need to be taken extremely seriously by workers in the field.
 Please wait while we load your content...
Please wait while we load your content...

