
Ferroelastic property of tetramethylammonium tetrachlorozincate tetrachlorocuprate, [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, 0.5, and 1)
Ae Ran Lim*ab
aDepartment of Science Education, Jeonju University, Jeonju 560-759, Korea. E-mail: aeranlim@hanmail.net; arlim@jj.ac.kr; Fax: +82-63-220-2053; Tel: +82-63-220-2514
bDepartment of Carbon Fusion Engineering, Jeonju University, Jeonju 560-759, Korea
First published on 10th March 2015
Abstract
The various crystallographic structures of [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, 0.5, and 1) may be understood by considering the different chemical shifts observed in 1H MAS and 13C CP/MAS NMR spectra. Cu2+ ions, after replacing partially the Zn2+ ions, occupy the same locations in the lattice as the Zn2+ ions. The NMR spectrum and T1ρ of x = 0.1 and 0.3 were found to be similar to those of pure [N(CH3)4]2ZnCl4, whereas the NMR spectrum and T1ρ of x = 0.5 were different. Consequently, the existence of ferroelastic properties of N(CH3)4 ions in x = 0, 0.1, 0.3, and 1 are apparent at low temperatures, whereas they disappear for x = 0.5. It has been demonstrated that the replacement of Zn2+ ions with high concentrations of Cu2+ ions changes the ferroelastic property of the crystal.
I. Introduction
Crystals of the formula A2BX4 have received a great deal of attention owing to their interesting phase transition sequences. [N(CH3)4]2ZnCl4 and [N(CH3)4]2CuCl4 single crystals are A2BX4-type crystals. Tetramethylammonium tetrachlorozincate, [N(CH3)4]2ZnCl4, undergoes five phase transitions at the following temperatures: 161 K (TC5), 181 K (TC4), 276.3 K (TC3), 279 K (TC2), and 296 K (TC1).1–7 These previous studies have concluded that there are six phases, I to VI in the order of decreasing temperature, of [N(CH3)4]2ZnCl4 crystals. The crystal structure of phase I is orthorhombic. The transition from the normal (I) to the incommensurate (II) phase occurs at 296 K. The corresponding symmetry changes are as follows: the ferroelectric phase III is orthorhombic with space group P21cn; the ferroelastic phase IV is monoclinic with space group P21/n; the ferroelastic phase V is monoclinic with space group P21/c; and finally, phase VI is orthorhombic with space group P212121.5 Furthermore, tetramethylammonium tetrachlorocuprate, [N(CH3)4]2CuCl4, crystals exhibit four phases, I to IV, with transition temperatures of 263 K (TC3), 291 K (TC2), and 301 K (TC1).8 The structure and space group of the lowest temperature phase IV is monoclinic with space group P1121/n. The structure of phase III is monoclinic with space group P121/c1, and phase II is incommensurate. The highest temperature phase, phase I, has an orthorhombic structure with space group Pmcn. These three phase transitions in [N(CH3)4]2CuCl4 are, in the order of increasing temperature: ferroelastic (IV)–ferroelastic (III), ferroelastic (III)–incommensurate (II), and incommensurate (II)–commensurate (I).9 In particular, these two compounds, [N(CH3)4]2ZnCl4 and [N(CH3)4]2CuCl4, have the ferroelastic property at low temperatures.Ferroelasticity was first recognized as a structure property by Aizu in 1970.10 A crystal is ferroelastic if it has two or more stable orientation states in the absence of mechanical stress, and can be reversibly transformed from one to another of these states by the application of mechanical stress. When a ferroelastic crystal is heated, the ferroelastic effect usually disappears at a well-defined temperature. At this temperature, a structural phase transition occurs between a ferroelastic and a paraelastic phase; the main feature of which is that a ferroelastic hysteresis exists in one phase but not in the other. Also, the ferroelastic domain occurs in all ferroelastic crystals as a consequence of the reduction in symmetry between the paraelastic and ferroelastic phases.
The physical properties of [N(CH3)4]2ZnCl4 and [N(CH3)4]2CuCl4 have been studied using various experimental methods by several research groups.11–20 Ribet et al.21 reported observing ferroelectric–ferroelastic phase transitions of [N(CH3)4]2ZnCl4 through X-ray and synchrotron topography. Recently, the roles of chemically inequivalent a-N(CH3)4 and b-N(CH3)4 ions in [N(CH3)4]2ZnCl4 and [N(CH3)4]2CuCl4 have been reported by static nuclear magnetic resonance (NMR) and magic angle spinning (MAS) NMR, respectively.22,23 Furthermore, the ferroelastic phase transition of [N(CH3)4]2ZnCl4 and [N(CH3)4]2CuCl4 at low temperatures have been discussed.24
In this work, [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, 0.5, and 1) single crystals were grown from aqueous solutions by the slow evaporation method. We measured the temperature dependences for 1H magic angle spinning (MAS) NMR spectrum and 13C cross-polarization (CP)/MAS NMR spectrum of [N(CH3)4]2Zn1−xCuxCl4 to elucidate the structural geometry. In addition, we determined the spin–lattice relaxation times in the rotating frame, T1ρ, for 1H and 13C nuclei in [N(CH3)4]2Zn1−xCuxCl4, for varying amounts of impurity Cu2+ ions. This is the first time that the local structures of [N(CH3)4]2Zn1−xCuxCl4 have been investigated, and we used the results to analyze the role of N(CH3)4 ions. These results enabled us to compare the structural properties of pure [N(CH3)4]2ZnCl4 and [N(CH3)4]2CuCl4, and examine the effect of substituting Zn2+ ions in [N(CH3)4]2ZnCl4 with Cu2+ ions, with a focus on the effects of such substitution on ferroelasticity. Furthermore, in order to confirm the ferroelastic properties, domain structures were observed using an optical polarizing microscope.
II. Crystal structure
At room temperature, the [N(CH3)4]2Zn0.5Cu0.5Cl4 crystal is an orthorhombic system (space group P21cn) with Z = 4 and the following unit cell dimensions: a = 8.988 Å, b = 15.527 Å, and c = 12.269 Å.25 The atomic arrangement in [N(CH3)4]2Zn0.5Cu0.5Cl4 consists of alternate organic–inorganic layers of a-N(CH3)4/Zn(Cu)Cl4 and organic sheets b-N(CH3)4, both parallel to b-plane (Fig. 1).26 This figure shows chains that are repeated sequences of Cu(Zn)Cl4 and a-N(CH3)4, where the sense of tetrahedron orientation alternates within the same chain. The b-N(CH3)4 tetrahedra are present between the chains and on both sides of the organic–inorganic layers, building organic layers. | ||
| Fig. 1 The structure of [N(CH3)4]2Zn0.5Cu0.5Cl4 in the ab-plane. Cu/ZnCl42− anions are represented by grey tetrahedrons. N(CH3)4+ cations are represented by empty tetrahedrons. The dashed boundaries show the development of the zigzag chain in the organic–inorganic layer along the [110] direction. | ||
A [N(CH3)4]2ZnCl4 crystal in phase I has an orthorhombic structure with space group Pmcn. Its orthorhombic lattice constants are: a = 8.946 Å, b = 15.515 Å, and c = 12.268 Å.27,28 In this phase, a unit cell contains Z = 4 units consisting of two inequivalent kinds of tetramethylammonium ions, hereafter abbreviated as a-N(CH3)4 and b-N(CH3)4, and one kind of ZnCl42− ion.27 The ZnCl4 ion and the a-N(CH3)4 are positioned in a strongly correlated manner, while the b-N(CH3)4 is less correlated than the other kind of ions. Moreover, a [N(CH3)4]2CuCl4 crystal in phase I has an orthorhombic structure, and its orthorhombic lattice constants are: a = 9.039 Å, b = 15.515 Å, and c = 12.268 Å, which are slightly different from those for the hexagonal form.29,30 In this phase, a unit cell contains Z = 4 units consisting of two inequivalent kinds of tetramethylammonium ions, as well as [N(CH3)4]2ZnCl4.
III. Experimental method
[N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, 0.5, and 1) single crystals were grown at room temperature by slow evaporation of an aqueous solution containing ZnCl2, CuCl2, and N(CH3)4Cl, in stoichiometric proportions. [N(CH3)4]2Zn1−xCuxCl4 single crystals varied in color according to the amount of Cu2+ ions, as shown in Fig. 2. | ||
| Fig. 2 The colors of mixed crystals [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, 0.5, and 1). | ||
Solid-state NMR experiments were performed using a Bruker DSX 400 FT NMR spectrometer at the Korea Basic Science Institute. 1H MAS NMR and 13C CP/MAS NMR experiments were performed at the Larmor frequencies of 400.12 MHz and 100.61 MHz, respectively. The samples were placed in the 4 mm CP/MAS probe as powders. The MAS rate was set to 10 kHz and 7 kHz for 1H MAS and 13C CP/MAS, respectively, to minimize the spinning sideband overlap. Here, the frequency scale of the spectrum for 1H and 13C was expressed with respect to tetramethylsilane (TMS). In the case for 1H, the T1ρ measurements were performed using π/2–t–acquisition. The spin–lattice relaxation times in the rotating frame, T1ρ, were measured by varying the length of the spin-locking pulses. The π/2 pulse width used for T1ρ was 5 μs, corresponding to the frequency of the spin-locking field, 50 kHz. Moreover, the T1ρ for 13C was obtained using CP–t–acquisition, and the frequency of the spin-locking field was 78.1 kHz. The experimental temperatures were maintained at constant values, with an accuracy of ±0.5 K, by controlling the nitrogen gas flow and heater current. The temperature-dependent NMR measurements were carried out in the temperature range from 150 to 450 K.
IV. Experimental results and analysis
At room temperature, the structures of the [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, 0.5, and 1) crystals were determined with an X-ray diffraction system (Bruker AXS GMBH) at the Korea Basic Science Institute. The single crystals were mounted on a Bruker SMART CCD diffractometer equipped with a graphite-monochromated Mo Kα (λ = 0.71073 Å) radiation source. Data collection and integration were performed at 298 K with SMART (Bruker, 2000) and SAINT-Plus (Bruker, 2001).31 The lattice constants of the five crystals were shown in Table 1, and all the [N(CH3)4]2Zn1−xCuxCl4 crystals containing Cu2+ impurities had the same orthorhombic structure as [N(CH3)4]2ZnCl4 and [N(CH3)4]2CuCl4. And, the chemical composition of the crystals was confirmed with an electron probe microanalyzer (EPMA 1600). The X-ray diffraction and elemental analysis data indicate that these single crystals are [N(CH3)4]2Zn1−xCuxCl4 (x = 0.1, 0.3, and 0.5). In addition, in order to determine the phase transition temperatures, differential scanning calorimetry (DSC) was carried out on the crystals with a Dupont 2010 DSC instrument. The measurements were performed at a heating rate of 10 °C min−1 in the temperature range from 200 K to 500 K, and the endothermic peaks for x = 0.1, 0.3, and 0.5 were shown in Fig. 3. The phase transition temperatures are nearly unchanged with varying amounts of impurity Cu2+ ions and are similar to those for pure [N(CH3)4]2ZnCl4.| a | b | c | |
|---|---|---|---|
| [N(CH3)4]2ZnCl4 (x = 0) | 8.9958 ± 0.0030 | 15.5162 ± 0.0039 | 12.2517 ± 0.0034 |
| [N(CH3)4]2Zn0.9Cu0.1Cl4 (x = 0.1) | 8.9994 ± 0.0027 | 15.5164 ± 0.0037 | 12.2713 ± 0.0029 |
| [N(CH3)4]2Zn0.7Cu0.3Cl4 (x = 0.3) | 8.9988 ± 0.0020 | 15.5388 ± 0.0035 | 12.2663 ± 0.0035 |
| [N(CH3)4]2Zn0.5Cu0.5Cl4 (x = 0.5) | 9.0068 ± 0.0027 | 15.5409 ± 0.0043 | 12.2774 ± 0.0032 |
| [N(CH3)4]2CuCl4 (x = 1) | 9.1136 ± 0.0029 | 15.2723 ± 0.0046 | 12.1526 ± 0.0050 |
 | ||
| Fig. 3 Differential scanning calorimetry (DSC) thermogram of [N(CH3)4]2Zn1−xCuxCl4 (x = 0.1, 0.3, and 0.5) single crystals. | ||
A. 1H MAS NMR in [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, 0.5, and 1)
Structural analysis of [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, 0.5, and 1) was carried out with the 1H MAS NMR method. The chemical shifts for 1H in [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, and 0.5) were measured over the temperature range of 180–425 K, as shown in Fig. 4(a). In the cases of x = 0, 0.1, 0.3, and 0.5, the small changes in the chemical shifts near 296 K correspond to phase transitions. The chemical shift of [N(CH3)4]2Zn0.9Cu0.1Cl4 with x = 0.1 changes near 276 K, meaning structural phase transition. However, the other phase transitions cannot be identified from the chemical shifts. Alternatively, the insert in Fig. 4(b) shows the 1H MAS NMR spectrum of [N(CH3)4]2CuCl4 with x = 1 at room temperature. The NMR spectrum consists of two peaks at chemical shifts of 2.45 and 6.80 ppm. The spinning sidebands are marked with asterisks. The signals at chemical shifts of 2.45 and 6.80 ppm are assigned to the methyl protons, and they are clearly due to magnetically inequivalent sites. Here, the two proton peaks at 2.45 and 6.80 ppm cannot be distinguished between a-N(CH3)4 or b-N(CH3)4. The 1H chemical shift changes with increasing temperature, as shown in Fig. 4(b). The chemical shifts near TC3 also change abruptly, whereas those near TC1 and TC2 change almost continuously. The chemical shift in the case for x = 1 is completely different from those for x = 0, 0.1, 0.3, and 0.5. This difference is due to variations in the electronic structure of the Zn2+ and Cu2+ ions. | ||
| Fig. 4 (a) Chemical shifts of the 1H MAS NMR spectrum as a function of temperature in [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, and 0.5) and (b) chemical shift of the 1H MAS NMR spectrum as a function of temperature in [N(CH3)4]2CuCl4 (x = 1) (inset: 1H MAS NMR spectrum for [N(CH3)4]2CuCl4 at room temperature). | ||
The spin–lattice relaxation times in the rotating frame, T1ρ, for the proton in [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.3, 0.5, and 1) were obtained as a function of temperature. The nuclear magnetization recovery curves obtained for protons were described by the following single exponential function:32–34 S(t) = S0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/T1ρ), where S(t) is the magnetization at time, t, and S0 is the total nuclear magnetization of 1H at thermal equilibrium. The slopes of the recovery traces are different at each temperature. The temperature dependences of the 1H T1ρ are shown in Fig. 5. When the paramagnetic Cu2+ impurity was included, x = 0.3 in [N(CH3)4]2Zn1−xCuxCl4, the trend in T1ρ resembles that of 1H T1ρ in pure [N(CH3)4]2ZnCl4. Below TC3, T1ρ increases abruptly, and the proton T1ρ data does not show any evidence of an anomalous change near the phase transition temperatures of TC1 and TC2. However, the 1H T1ρ curve of [N(CH3)4]2Zn0.5Cu0.5Cl4 is markedly different from those observed for pure [N(CH3)4]2ZnCl4 and [N(CH3)4]2CuCl4. Here, the 1H T1ρ values for [N(CH3)4]2Zn0.5Cu0.5Cl4 are smaller than those in [N(CH3)4]2ZnCl4, and larger than that in [N(CH3)4]2CuCl4. In the case for [N(CH3)4]2CuCl4 with x = 1, the 1H T1ρ was found to be much shorter than that for [N(CH3)4]2ZnCl4. The T1ρ values increase with increasing temperature and the change is discontinuous near TC3, but relatively continuous near TC1 and TC2.
exp(−t/T1ρ), where S(t) is the magnetization at time, t, and S0 is the total nuclear magnetization of 1H at thermal equilibrium. The slopes of the recovery traces are different at each temperature. The temperature dependences of the 1H T1ρ are shown in Fig. 5. When the paramagnetic Cu2+ impurity was included, x = 0.3 in [N(CH3)4]2Zn1−xCuxCl4, the trend in T1ρ resembles that of 1H T1ρ in pure [N(CH3)4]2ZnCl4. Below TC3, T1ρ increases abruptly, and the proton T1ρ data does not show any evidence of an anomalous change near the phase transition temperatures of TC1 and TC2. However, the 1H T1ρ curve of [N(CH3)4]2Zn0.5Cu0.5Cl4 is markedly different from those observed for pure [N(CH3)4]2ZnCl4 and [N(CH3)4]2CuCl4. Here, the 1H T1ρ values for [N(CH3)4]2Zn0.5Cu0.5Cl4 are smaller than those in [N(CH3)4]2ZnCl4, and larger than that in [N(CH3)4]2CuCl4. In the case for [N(CH3)4]2CuCl4 with x = 1, the 1H T1ρ was found to be much shorter than that for [N(CH3)4]2ZnCl4. The T1ρ values increase with increasing temperature and the change is discontinuous near TC3, but relatively continuous near TC1 and TC2.
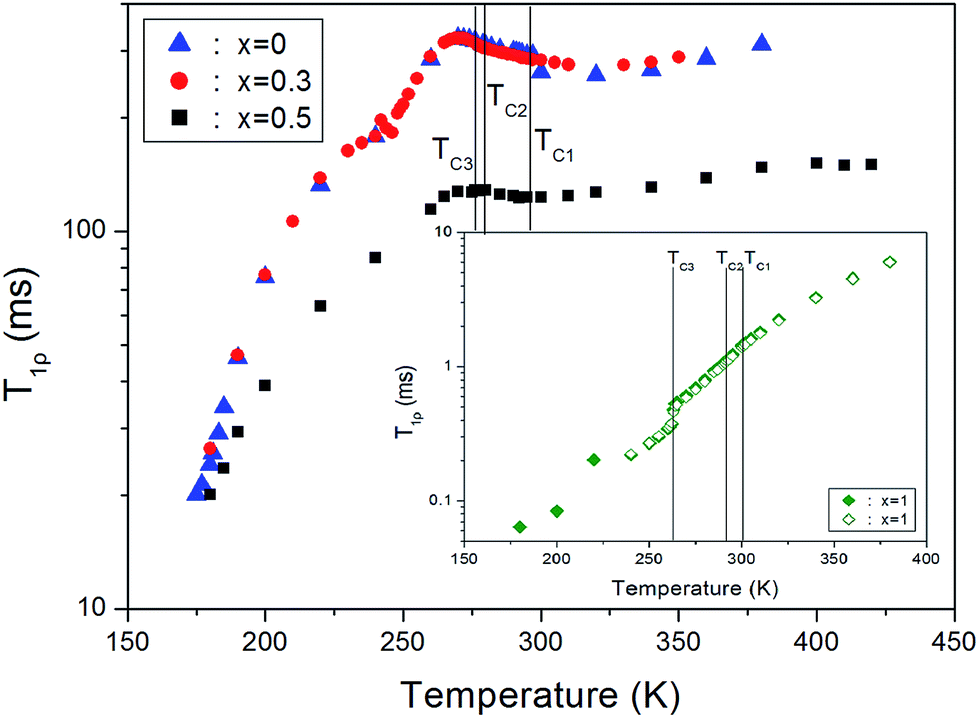 | ||
| Fig. 5 Temperature dependences of the 1H spin–lattice relaxation time in the rotating frame, T1ρ, in [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.3, and 0.5). (Inset: temperature dependences of the 1H spin–lattice relaxation time in the rotating frame, T1ρ, in [N(CH3)4]2CuCl4.) | ||
B. 13C CP/MAS NMR in [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, 0.5, and 1)
Structural analysis of [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, 0.5, and 1) was carried out using 13C NMR spectroscopy. The chemical shifts for 13C in [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, and 0.5) were measured over the temperature range of 170–430 K, as shown in Fig. 6(a). In the cases for x = 0, 0.1, 0.3, and 0.5, the 13C CP/MAS NMR spectrum in the temperature range 293–380 K consists of a single resonance line for one type of N(CH3)4, as shown in Fig. 6(a). The chemical shifts of CH3 in the two inequivalent kinds of a-N(CH3)4 and b-N(CH3)4 in [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, and 0.5) were not measured within this temperature range. In the case for [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, and 0.3), the 13C NMR chemical shift at the transition point of 276 K (=TC3) splits into two lines, as shown in Fig. 6(a). This splitting indicates that at this temperature there is a phase transition to a new phase with a monoclinic symmetry lower than the orthorhombic symmetry. The III–IV transition results in an abrupt splitting of the 13C NMR line into two components, indicative of a ferroelastic property. The ferroelastic domain structures in phase IV of [N(CH3)4]2ZnCl4 (x = 0) and [N(CH3)4]2Zn0.7Cu0.3Cl4 (x = 0.3) were confirmed by employing an optical polarizing microscope. Fig. 6(c) shows the domain patterns for the ferroelastic and paraelastic phases of x = 0 and x = 0.3. The domain patterns at 350 K do not appear similar to those for the paraelectric phase. The appearance of microscope domain walls with many parallel lines with decreasing temperature is a property of the ferroelastic phase. In the case for [N(CH3)4]2Zn0.5Cu0.5Cl4 (x = 0.5), the 13C NMR spectrum at all temperatures measured here consists of only one resonance line, as shown in Fig. 6(a). The in situ 13C CP/MAS NMR spectrum for [N(CH3)4]2Zn0.5Cu0.5Cl4 are also shown in Fig. 6(b) as a function of temperature. Furthermore, there were only continuous quantitative changes in the chemical shift, where the 13C chemical shift slowly and monotonically increases with temperature. In the case for [N(CH3)4]2Zn0.5Cu0.5Cl4 with x = 0.5 the domain walls did not appear at all temperatures, as shown in Fig. 6(c).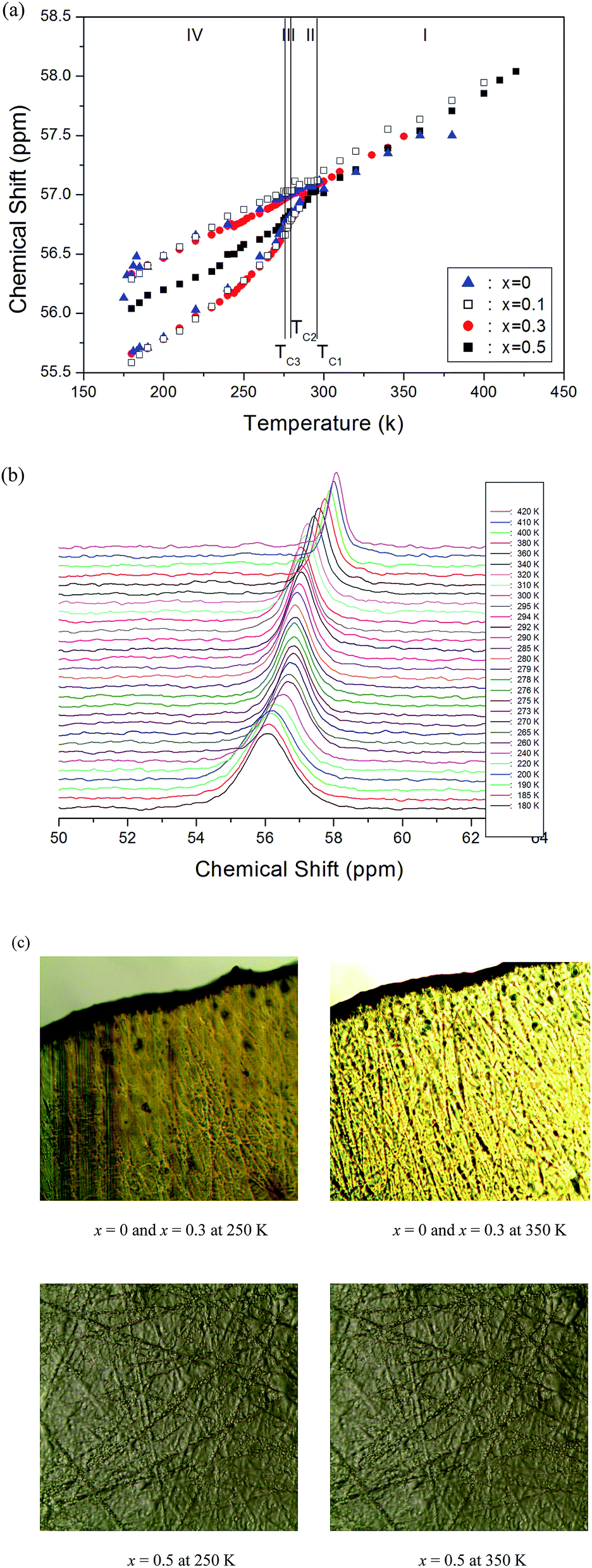 | ||
| Fig. 6 (a) Chemical shifts of the 13C CP/MAS NMR spectrum as a function of temperature in [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, and 0.5), (b) in situ 13C CP/MAS NMR spectrum as a function of temperature in [N(CH3)4]2Zn0.5Cu0.5Cl4, and (c) domain wall patterns of [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.3, and 0.5) at 250 K and 350 K obtained with optical polarizing microscopy. | ||
On the other hand, the 13C CP/MAS NMR spectrum for [N(CH3)4]2CuCl4 at room temperature has two signals at chemical shifts of δ = 72.13 and 133.48 ppm. The signals at chemical shifts of δ = 72.13 and 133.48 ppm represent the methyl carbons in inequivalent b-N(CH3)4 and a-N(CH3)4, respectively. In the X-ray diffraction study of Hasebe et al.,27 the deformation of the b-N(CH3)4 ion was larger than that for the a-N(CH3)4 ion, and the degree of the deformation was enlarged in the ferroelectric phase. Based on these results, a-N(CH3)4 and b-N(CH3)4 are defined according the change of the relaxation time as a function of temperature, which is discussed next. Fig. 7(a) shows the 13C CP/MAS NMR spectrum below 291 K, in which the 13C chemical shifts are shown by the three lines. Above TC2 (291 K), the 13C NMR spectrum consists of two lines for a-N(CH3)4 and b-N(CH3)4, as shown in Fig. 7(a). However, at the transition point at 291 K, the 13C NMR chemical shifts split into three lines. This splitting indicates that there is a phase transition at this temperature to a new phase with monoclinic symmetry, which is a symmetry reduction from orthorhombic symmetry. Thus, the II–III transition results in an abrupt splitting of the 13C NMR line into three components, which is indicative of ferroelasticity. Furthermore, above 291 K, there were only continuous quantitative changes in the chemical shift, where the 13C chemical shift slowly and monotonically decreases with increasing temperature. The ferroelastic domain structures in phases III and IV of [N(CH3)4]2CuCl4 with x = 1 were observed by employing an optical polarizing microscope, as shown in Fig. 7(b). The domain walls in the ferroelastic phase of phase IV were measured, whereas those in the paraelastic phase of phase I had disappeared. Consequently, the NMR spectrum and domain walls of [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, and 1) below 276 K show the ferroelastic characteristic, whereas the NMR spectrum and domain patterns of [N(CH3)4]2Zn0.5Cu0.5Cl4 below 276 K do not.
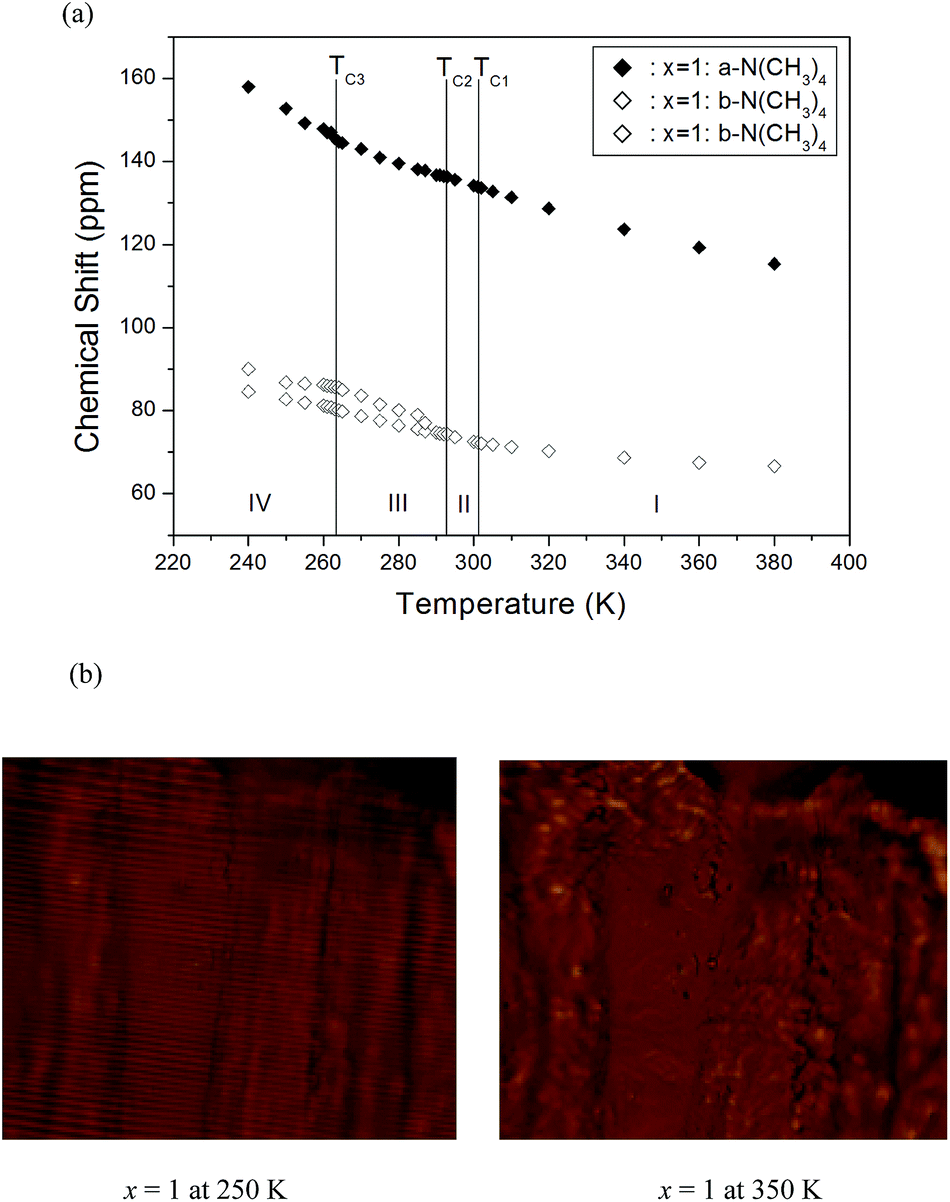 | ||
| Fig. 7 (a) Chemical shift of the 13C CP/MAS NMR spectrum as a function of temperature in [N(CH3)4]2CuCl4 (x = 1) and (b) domain wall patterns of [N(CH3)4]2CuCl4 at 250 K and 350 K obtained with optical polarizing microscopy. | ||
The spin–lattice relaxation times in the rotating frame, T1ρ, in the [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.3, 0.5, and 1) were obtained for each carbon as a function of temperature, with variable spin locks on the carbon channel following cross-polarization. The 13C magnetization was generated by cross-polarization, after spin locking of the protons. The proton field was then turned off for a variable time, t, while the 13C rf field remained on. Finally, the 13C free induction decay was observed under high-power proton decoupling, and a Fourier transform was subsequently applied. Values of T1ρ could be selected by the Fourier transformation of the free-induction decay (FID), after spin locking and repetition of the experiment with variations in the time, t. The signals obtained for carbon were described by a single exponential function. Fig. 8(a) shows the T1ρ values for 13C in the cases of x = 0, 0.3, and 0.5. The slopes of the T1ρ values near 296 K (=TC1) are different, and this temperature corresponds to phase transition. The T1ρ values for two 13C signals in the ferroelastic phase are nearly the same within the experimental error range. In the case for [N(CH3)4]2CuCl4 with x = 1, the 13C T1ρ values for a-N(CH3)4 and b-N(CH3)4, shown in Fig. 8(b), are similar, especially at higher temperatures. However, near TC3, the T1ρ values change abruptly, and the T1ρ values for the two 13C signals of b-N(CH3)4, produced by the ferroelastic twin structure below TC2, are the same within the experimental error range.
 | ||
| Fig. 8 (a) Temperature dependences of the 13C spin–lattice relaxation time in the rotating frame, T1ρ, in [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.3, and 0.5) and (b) temperature dependences of the 13C T1ρ in [N(CH3)4]2CuCl4. | ||
V. Discussion
The structures and the phase transition temperatures of the mixed crystals [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, 0.5, and 1) were determined with X-ray diffraction and DSC, respectively. Here, the structure or phase transition temperatures were almost unchanged by the doping of [N(CH3)4]2ZnCl4 crystals with Cu2+ ions.The chemical shifts for 1H and 13C nuclei in [N(CH3)4]2Zn1−xCuxCl4 were studied as a function of temperature. The chemical shifts in [N(CH3)4]2Zn1−xCuxCl4 varied according to the concentration of Cu2+ ions. The differences in the chemical shifts among the members of the series could potentially be due to differences in the electron structures of Zn2+ and Cu2+, in particular, the structure of the d electrons, which screen the nuclear charge from the motion of the outer electrons. Zn2+ has a filled d shell, whereas Cu2+ has one s electron outside the closed d shell.
[N(CH3)4]2ZnCl4 and [N(CH3)4]2CuCl4 contains two inequivalent types of N(CH3)4 ions, a-N(CH3)4 and b-N(CH3)4, respectively. Here, the two inequivalent kinds of a-N(CH3)4 and b-N(CH3)4 in [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, and 0.5) were not measured. However, the two crystallographically different ions a-N(CH3)4 and b-N(CH3)4 in [N(CH3)4]2CuCl4 were identified using 13C CP/MAS NMR. However, the 1H and 13C T1ρ values were obtained with varying concentrations of Cu2+ ions in [N(CH3)4]2Zn1−xCuxCl4. It is apparent that T1ρ for 1H and 13C are not governed by the same mechanism for the amount of paramagnetic impurity Cu2+. As a result, the trends in the NMR spectrum and T1ρ of 1H and 13C nuclei in the [N(CH3)4]2Zn1−xCuxCl4 (x = 0.1 and 0.3) were similar to those for [N(CH3)4]2ZnCl4. However, the structural properties of [N(CH3)4]2Zn0.5Cu0.5Cl4 were strongly affected.
The T1ρ values of materials including paramagnetic ions are smaller than those of pure [N(CH3)4]2ZnCl4. Because T1ρ should be inversely proportional to the concentration and to the square of the magnetic moment of the paramagnetic ions, the T1ρ values for samples containing paramagnetic ions are generally smaller than for those without. Therefore, T1ρ for 1H and 13C are driven in these systems by the fluctuations of the magnetic dipole of the Cu2+ paramagnetic ions.
VI. Conclusion
The purpose of this study was to investigate how the local structure in a pure crystal is affected by the random presence of a cation of a different size, and to determine the influence of this substitution on the physical properties of the crystal. After the partial replacement of Zn2+ ions by Cu2+ ions, the Cu2+ ions occupied the same locations in the lattice as the Zn2+ ions did. Their crystallographic structures in [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, 0.5, and 1) can be understood by considering the differences in the chemical shifts of the 1H MAS NMR and 13C CP/MAS NMR spectra. The NMR spectrum and T1ρ for x = 0.1 and 0.3 were similar to those for pure [N(CH3)4]2ZnCl4, whereas the NMR spectrum and T1ρ for x = 0.5 were different.The variation of the structural geometry, as a function of impurity concentration in the mixed system, was interpreted in terms of the differences in size and electron structure between the host and impurity ions. In particular, we attempted to explain the role of CH3 in the spin–lattice relaxation time mechanisms for the systems containing the paramagnetic Cu2+ impurity, based on the 1H MAS NMR and 13C CP/MAS NMR data in [N(CH3)4]2ZnCl4 and [N(CH3)4]2CuCl4. Consequently, the existence of ferroelastic properties of N(CH3)4 ions in [N(CH3)4]2Zn1−xCuxCl4 (x = 0, 0.1, 0.3, and 1) were apparent at low temperatures, whereas they were absent for [N(CH3)4]2Zn0.5Cu0.5Cl4. This study has shown that the replacement of Zn2+ ions with a high concentration of Cu2+ ions causes the ferroelastic property of the structure to disappear.
Acknowledgements
This research was supported by the Basic Science Research program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (2012001763).References
- J. D. Axe, Incommensurate phases in Dielectrics, ed. R. Blinc and A. P. Levanyuk, North-Holland, Amsterdam, 1986 Search PubMed.
- H. Z. Cummins, Phys. Rep., 1990, 185, 211 CrossRef CAS.
- I. R. Larrea, A. Lopez Echarri and M. J. Tello, J. Phys. C: Solid State Phys., 1981, 14, 3171 CrossRef.
- M. Hohioka, J. Phys. Soc. Jpn., 1983, 52, 4056 CrossRef.
- S. Sawada, I. Yamaguchi, H. Suzuki and F. Shimizu, J. Phys. Soc. Jpn., 1985, 54, 3129 CrossRef CAS.
- K. Parlinski and F. Denoyer, J. Phys. C: Solid State Phys., 1985, 18, 293 CrossRef CAS.
- G. Madariaga, F. J. Zuniga, J. M. Perez-Mato and M. J. Tello, Acta Crystallogr., Sect. B: Struct. Sci., 1987, 43, 356 CrossRef.
- B. Martin, J. M. Pastar, F. Rull and J. A. De Saja, Solid State Commun., 1982, 44, 1047 CrossRef CAS.
- A. G. Cuevas, M. J. Tello, J. Fernandez, A. L. Echarri, J. Herreros and M. Couzi, J. Phys. C: Solid State Phys., 1983, 16, 473 CrossRef.
- K. Aizu, Phys. Rev. B: Solid State, 1970, 2, 754 CrossRef.
- A. A. El Fadl, A. El-Korashy and H. El-Zahed, J. Phys. Chem. Solids, 2002, 63, 375 CrossRef CAS.
- A. El-Korashy, J. Mater. Sci. Lett., 2000, 19, 29 CrossRef CAS.
- V. B. Kapustyanyk and A. Y. Batyuk, J. Appl. Spectrosc., 2004, 71, 387 CrossRef CAS.
- S. A. Sveleba, I. V. Karpa, I. M. Katerynchuk, O. V. Semotyuk, R. M. Shymkiv, I. M. Kunyo and O. I. Fitsych, J. Appl. Spectrosc., 2011, 78, 228 CrossRef CAS.
- D. G. Sannikov, Phys. Solid State, 2005, 47, 324 CrossRef CAS.
- D. G. Sannikov and G. A. Kessenikh, Phys. Solid State, 2005, 47, 729 CrossRef CAS.
- D. G. Sannikov, Phys. Solid State, 2000, 42, 2282 CrossRef CAS.
- A. Y. Batyuk, V. B. Kapustyanyk and Y. M. Korchak, J. Appl. Spectrosc., 2005, 72, 413 CrossRef CAS PubMed.
- R. M. Shymkiv, S. A. Sveleba, I. V. Karpa, I. N. Katerynchuk, I. M. Kunyo and E. I. Phitsych, J. Appl. Spectrosc., 2011, 78, 823 CrossRef PubMed.
- A. El-Korashy and M. G. Brik, Solid State Commun., 2005, 135, 298 CrossRef CAS PubMed.
- M. Ribet, S. Leon, F. Lefaucheux and M. C. Robert, J. Appl. Crystallogr., 1990, 23, 277 CrossRef CAS.
- A. R. Lim and K.-Y. Lim, Solid State Sci., 2014, 31, 70 CrossRef CAS PubMed.
- A. R. Lim, J. Mol. Struct., 2014, 1056–1057, 233 CrossRef CAS PubMed.
- A. R. Lim, D. K. Park, J.-H. Chang and S.-Y. Jeong, J. Phys. Soc. Jpn., 2001, 70, 1937 CrossRef CAS.
- A. B. Rhaiem, F. Hiel, K. Geidara and M. Gargouri, Spectrochim. Acta, Part A, 2007, 66, 1107 CrossRef PubMed.
- F. Hlel, A. B. Rhaiem and K. Guidara, Russ. J. Inorg. Chem., 2008, 53, 785 CrossRef.
- K. Hasebe, H. Mashiyama, N. Koshiji and S. Tanisaki, J. Phys. Soc. Jpn., 1987, 56, 3543 CrossRef CAS.
- F. J. Zuniga, G. Madariaga and J. M. Perez-Mato, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45, 462 CrossRef.
- W. Rehwald and A. Vonlanthen, Z. Phys. B: Condens. Matter, 1985, 61, 25 CrossRef CAS.
- K. Gesi and M. Iizumi, J. Phys. Soc. Jpn., 1980, 48, 1775 CrossRef CAS.
- SMART and SAINT-Plus v6.22, Bruker AXS Inc., Madison, Wisconsin, USA, 2000 Search PubMed.
- J. L. Koenig, Spectroscopy of Polymers, Elsevier, New York, 1999 Search PubMed.
- B. Cowan, Nuclear Magnetic Resonance and Relaxation, Cambridge Univ., UK, 1997 Search PubMed.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Phys. Rev., 1948, 73, 679 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |