
Synthesis of dual type Fe species supported mesostructured silica nanoparticles: synergistical effects in photocatalytic activity†
Abstract
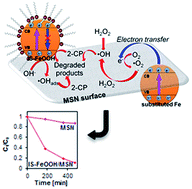
Dual type Fe species (isomorphously substituted Fe species and a colloidal α-FeOOH (IS-FeOOH)) supported on mesostructured silica nanoparticles (IS-FeOOH/MSN) were prepared by a simple electrochemical method followed by impregnation. Characterization was conducted using X-ray diffraction, transmission electron microscopy, surface area analysis, Fourier-transform infrared spectroscopy, nuclear magnetic resonance, electron spin resonance, and X-ray photoelectron spectroscopy. The results suggested that silica removal occurred in the MSN framework to isomorphously substitute Fe cations while retaining the colloidal structure of IS-FeOOH. The catalytic activity of IS-FeOOH/MSN was tested on photo-Fenton-like degradation of 2-chlorophenol under fluorescent light irradiation. The performance of the catalyst was in the following order: 10 wt% IS-FeOOH/MSN > 15 wt% IS-FeOOH/MSN > 5 wt% IS-FeOOH/MSN > MSN, with removal percentages of 92.2, 79.3, 73.1, and 14.2%, respectively. The results suggest that a synergistic effect between the dual type of Fe species (Si–O–Fe and IS-FeOOH colloid) and MSN played important roles in enhancing the degradation. The results provide strong evidence to support the potential use of IS-FeOOH/MSN as a photo-Fenton-like nanocatalyst for organic pollutants treatment.
 Please wait while we load your content...
Please wait while we load your content...

